Title 40 CFR
Part 191
Subparts B and C
Compliance Recertification
Application
for the
Waste Isolation Pilot Plant
Appendix SOTERM-2009
Actinide Chemistry Source Term
United States
Department of Energy
Waste Isolation Pilot Plant
Carlsbad Field Office
Carlsbad, New Mexico
Appendix SOTERM-2009
Actinide Chemistry Source Term
SOTERM-2.0 Expected WIPP Repository Conditions, Chemistry, and Processes
SOTERM-2.1 Ambient Geochemical Conditions
SOTERM-2.2 Repository Conditions
SOTERM-2.2.1 Repository Pressure
SOTERM-2.2.2 Repository Temperature
SOTERM-2.2.3 Water Content and Relative Humidity
SOTERM-2.2.4 Minimum Repository Brine Volume
SOTERM-2.3 Repository Chemistry
SOTERM-2.3.2 Brine pH and pH Buffering
SOTERM-2.3.3 Selected MgO Chemistry and Reactions
SOTERM-2.3.4 Iron Chemistry and Corrosion
SOTERM-2.3.5 Chemistry of Lead in the WIPP
SOTERM-2.3.6 Organic Chelating Agents
SOTERM-2.3.7 CPR in WIPP Waste
SOTERM-2.4 Important Postemplacement Processes
SOTERM-2.4.1 Microbial Effects in the WIPP
SOTERM-2.4.1.1 Gas Generation and Microbial Degradation of CPR Materials
SOTERM-2.4.1.2 Bioreduction of Multivalent Actinides
SOTERM-2.4.2 Radiolysis Effects in the WIPP
SOTERM-2.4.2.1 Radiation Chemistry of Brine Systems
SOTERM-2.4.2.2 Potential Radiolytic Effects on Actinide Speciation and Solubility
SOTERM-2.5 Changes in WIPP Conditions since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.0 WIPP-Relevant Actinide Chemistry
SOTERM-3.1 Actinide Inventory in the WIPP
SOTERM-3.2.1 Thorium Environmental Chemistry
SOTERM-3.2.2 WIPP-Specific Results since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.3.1 Uranium Environmental Chemistry
SOTERM-3.3.1.1 Uranium Subsurface Redox Chemistry
SOTERM-3.3.1.2 Solubility of U(IV)
SOTERM-3.3.1.3 Solubility and Speciation of U(VI)
SOTERM-3.3.2 WIPP-Specific Results since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.4 Neptunium Chemistry
SOTERM-3.4.1 Neptunium Environmental Chemistry
SOTERM-3.4.2 WIPP-Specific Results since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.5 Plutonium Chemistry
SOTERM-3.5.1 Plutonium Environmental Chemistry
SOTERM-3.5.1.1 Importance of Redox for Plutonium Speciation
SOTERM-3.5.1.2 Bioreduction of Higher-Valent Plutonium
SOTERM-3.5.1.3 Thermodynamic Stability of Higher-Valent Plutonium: PuO2+x
SOTERM-3.5.2 WIPP-Specific Results since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.6 Americium and Curium Chemistry
SOTERM-3.6.1 Americium and Curium Environmental Chemistry
SOTERM-3.6.2 WIPP-Specific Results since the CRA-2004 and the CRA-2004 PABC
SOTERM-3.7 Complexation of Actinides by WIPP Organic Chelating Agents
SOTERM-3.8.1 Actinide Colloids in the Environment
SOTERM-3.8.2 WIPP-Specific Results since the CRA-2004 PABC
SOTERM-3.9 Changes in Actinide Speciation Information since the CRA-2004 and the CRA-2004 PABC
SOTERM-4.0 Calculation of the WIPP Actinide Source Term
SOTERM-4.1 Overview of WIPP Approach to Calculate Actinide Solubilities
SOTERM-4.2 Use of Oxidation-State-Invariant Analogs
SOTERM-4.3 Actinide Inventory and Oxidation State Distribution in the WIPP
SOTERM-4.4 Actinide Speciation Reactions Used in the FMT Model
SOTERM-4.4.1 The III Actinides: Pu(III), Am(III), Cm(III)
SOTERM-4.4.2 The IV Actinides: Th(IV), U(IV), Pu(IV), Np(IV)
SOTERM-4.4.3 The V Actinides: Np(V)
SOTERM-4.4.4 The VI Actinides: U(VI)
SOTERM-4.5 Calculations of Actinide Solubility Using the FMT Computer Code
SOTERM-4.5.1 Pitzer Approach for High-Ionic-Strength Brines
SOTERM-4.5.2 Calculated Actinide Solubilities
SOTERM-4.6 Calculation of the Effects of Organic Ligands on Actinide Solubility
SOTERM-4.7 Calculation of Colloidal Contribution to Actinide Solution Concentrations
SOTERM-5.0 Use of the Actinide Source Term in PA
SOTERM-5.1.1 Elements and Isotopes Modeled
SOTERM-5.1.2 Use of Brine End Members
SOTERM-5.1.3 Sampling of Uncertain Parameters
SOTERM-5.1.4 Combining the Transport of Dissolved and Colloidal Species in the Salado
SOTERM-5.2 Construction of the Source Term
SOTERM-5.3 Example Calculation of Actinide Solubility
SOTERM-5.4 Calculated Dissolved, Colloidal, and Total Actinide Solubilities
Figure SOTERM-14. Redox Potential for Some Am Redox Couples (Silva et al. 1995, p. 74)
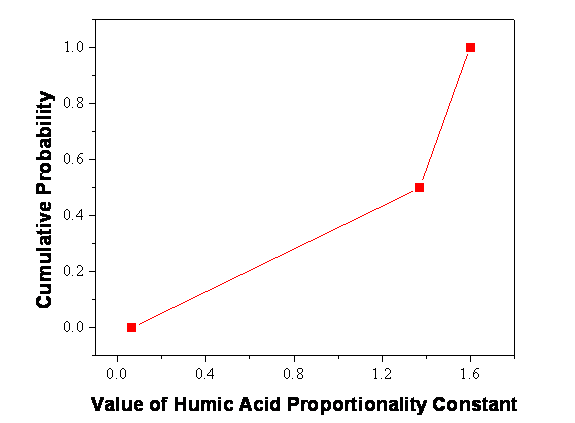
Figure SOTERM-21. Cumulative Distribution Function for the Humic-Acid Proportionality Constant
Table SOTERM-1. Summary of Current WIPP Chemistry Model Assumptions ( Leigh et al. 2005)
Table SOTERM-9. Thermodynamic Stability Constants for Key Th Hydrolytic Species
Table SOTERM-11. Solubility of U(VI) in High-Ionic-Strength Media
Table SOTERM-14. List of Documents and Reports that Support the CRA-2004 PABC
Table SOTERM-16. Oxidation States of the Actinides in the WIPP as Used in the CRA-2004 PABC
Table SOTERM-19. Comparison of Actinide Solubility Calculations With and Without Organics
Table SOTERM-20. Classification of Four Colloid Types Considered by WIPP PA
Table SOTERM-21. Material and Property Names for Colloidal Parameters
Table SOTERM-22. Colloid Concentration Factors (The CRA-2004, Appendix PA, Attachment SOTERM)
% percent
α alpha particle
ac activity of a chemical species
µm micrometer, micron
ACP LANL-CO Actinide Chemistry Project
am amorphous
AP Analysis Plan
aq aqueous
ASTP Actinide Source Term Program
atm atmosphere
β (apparent) stability constant, or beta particle
Bq becquerel
BRAGFLO Brine and Gas Flow code
CAPHUM maximum (cap) concentration of actinide associated with mobile humic colloids
CAPMIC maximum concentrations of actinides that could be associated with microbes
CCA Compliance Certification Application
Ci Curie
cm-1 per centimeter
CN coordination number
coll colloid
conc concentration
CONCINT concentration of actinide associated with mobile actinide intrinsic colloids
CONCMIN concentration of actinide associated with mobile mineral fragment colloids
CPR cellulosic, plastic, and rubber
CPu maximum concentration of all combined isotopes of Pu
cr crystalline phase
CRA Compliance Recertification Application
DBR direct brine release
D-H Debye-Hückel theory
DOE U.S. Department of Energy
DRZ disturbed rock zone
EDTA ethylenediaminetetraacetic acid
EPA U.S. Environmental Protection Agency
EQ3/6 software package for geochemical modeling of aqueous systems
eV electron volt
EXAFS Extended X-Ray Absorption Fine Structure
fCO2 fugacity of carbon dioxide
FMT Fracture-Matrix Transport
ft feet
g gamma radiation or activity coefficient
g gaseous, or gram
g/mL gram per milliliter
GBq giga becquerel
GWB Generic Weep Brine, a synthetic brine representative of fluids in Salado brine reservoirs
h hours
I ionic strength
K degree Kelvin or stability constant
kg kilogram
Ksp solubility product
L liter
LANL Los Alamos National Laboratory
LET Linear Energy Transfer
log logarithm
log10 logarithm base 10
m meter, molal
M mole per liter
m2 square meter
m3 cubic meter
mg/L milligram per liter
mL milliliter
mM millimole per liter
mol mole
molec molecule
MPa megapascal
N degree of polymerization number
NA Avogadro’s number
NIST National Institute of Standards and Technology
nm nanometer
NUTS Nuclide Transport System code
orgs organics
OXSTAT oxidation state parameter
PA Performance Assessment
PABC Performance Assessment Baseline Calculation
PAVT Performance Assessment Verification Test
pCH+ or pcH Negative logarithm of H+ concentration in moles per liter
pCO2 Partial pressure of carbon dioxide
pH negative logarithm of H+ activity
pHobs negative logarithm of H+ activity measured
PHUMCIM Proportionality constant for concentration of actinides associated with mobile humic colloids, in Castile brine
PHUMOXn Proportionality constant for humic colloids and actinides in the +n oxidation state
PHUMSIM Proportionality constant for concentration of actinides associated with mobile humic colloids, in Salado brine
pKa negative logarithm of the dissociation constant of an acid
pm picometer
pmH negative logarithm of H+ concentration in molal
PO2 partial pressure of molecular oxygen
PROPMIC proportionality constant describing the amount of actinide element bound to mobile microbes
RH relative humidity
s solid or second
SECOTP2D computer program that simulates single or multiple component radionuclide transport in fractures or granular aquifers
SEM scanning electron microscope
SNL Sandia National Laboratories
SOTERM WIPP Actinide Source Term
SPC Salado Primary Constituents
t½ half-life
TDS total dissolved solid
TRU transuranic
TWBIR Transuranic Waste Baseline Inventory Report
V volt, or vanadium
w with
WIPP Waste Isolation Pilot Plant
WWIS WIPP Waste Information System
XANES X-Ray Absorption Near Edge Structure
XRD X-Ray Diffraction
yr year
Am Americium
Am(III) Americium in the +3 oxidation state
Am(IV) Americium in the +4 oxidation state
Am(V) Americium in the +5 oxidation state
Am(VI) Americium in the +6 oxidation state
Am2+ Americium cation - Aqueous form of the americium in the +2 oxidation state that only exists as a transient
Am3+ Americium cation - Aqueous form of the americium in the +3 oxidation state
Am4+ Americium cation - Aqueous form of the americium in the +4 oxidation state
Am(CO3)n(3-2n) Americium (III) carbonate complex with n=1, 2, or 3
AmCO3OH Americium (III) carbonato hydroxide
AmO2+ Americium oxo-cation – Aqueous form of the americium in the +5 oxidation state
AmO22+ Americium oxo-cation – Aqueous form of the americium in the +6 oxidation state
AmO2OH Americium (V) oxide hydroxide
AmOH2+ Americium (III) hydroxide cation – (1:1) complex
Am(OH)2+ Americium (III) hydroxide cation – (1:2) complex
Am(OH)3 Americium hydroxide
AmPO4 Americium (III) phosphate
Am(SO4)n (3-2n) Americium (III) sulfate complex with n = 1 or 2
[An]p Concentration of an adsorbed actinide element (mol/particle)
An Actinide
An(III) General actinide in the +3 oxidation state
An(IV) General actinide in the +4 oxidation state
An(V) General actinide in the +5 oxidation state
An(VI) General actinide in the +6 oxidation state
An3+ Aqueous form of the actinide in the +3 oxidation state
An4+ Aqueous form of the actinide in the +4 oxidation state
Ann+ Aqueous form of the actinide in the +n oxidation state
An2(CO3)3 Actinide (III) carbonate – (2:3) complex
An2(CO3)22+ Actinide (III) carbonate iaon – (2:2) complex
AnB4O7+ Actinide (III) tetraborate ion – (1:1) complex
AnCl2+ Actinide (III) chloride ion – (1:1) complex
An(CO3)+ Actinide (III) carbonate ion – (1:1) complex
An(CO3)2- Actinide (III) carbonate ion – (1:2) complex
An(CO3)33- Actinide (III) carbonate ion – (1:3) complex
AnCO3OH Actinide (III) carbonate hydroxide
AnL (n+m) Complex of an actinide with a charge n and an organic ligand L with a charge m
An(V)O2+ or AnO2+ Aqueous form of the actinide in the +5 oxidation state
An(VI)O22+ or AnO22+ Aqueous form of the actinide in the +6 oxidation state
AnOH2+ Actinide (III) hydroxide cation – (1:1) complex
An(OH)3 Hydroxide of the actinide (III)
AnPO4 Actinide (III) phosphate
AnSO4+ Actinide (III) sulfate ion – (1:1) complex
B3O3(OH)4- Hydroxy polynuclear form of boric acid
B4O72- Tetraborate anion
B(OH)x3-x Hydroxyborate ions
Ba2+ Barium cation
Br- Bromide anion
[C] Concentration of species C in solution
[Cθ] Concentration of a chosen standard state
C Carbon or concentration
C6H10O5 Cellulose
CH4 Methane
CH3CO2- Acetate anion
(CH2CO2)2C(OH)(CO2) 3- Citrate anion
(CH2CO2)2N(CH2)2N(CH 2CO2)24- Ethylenediaminetetraacetate (EDTA) anion
C2O42- Oxalate anion
Ca2+ Calcium cation
CaCl2 Calcium chloride
CaCO3 Calcium carbonate
CaMg(CO3)2 Dolomite, calcium magnesium carbonate
CaO Calcium oxide
Ca4[Pu(OH)8]4+ Calcium plutonium (IV) hydroxide cation complex
CaSO4 Anhydrite, calcium sulfate
CaSO4×2H2O Gypsum, hydrated calcium sulfate
Ca4[Th(OH)8]4+ Calcium thorium (IV) hydroxide cation complex
CeO2 Cerium dioxide
Cl Chlorine
Cl- Chloride ion
Cl2 Chlorine
Cl2- Chlorine free radical
Cl3- Chlorine anion
ClBr- Chloride bromide radical
ClO- Hypochlorite anion
ClO2- Chlorite anion
ClO3- Chlorate anion
ClO4- Perchlorate anion
Cm Curium
Cm(III) Curium in the +3 oxidation state
Cm(IV) Curium in the +4 oxidation state
Cm3+ Curium cation – Aqueous form of the curium at the +3 oxidation state
CO2 Carbon dioxide
CO32- Carbonate anion
Cr Chromium
Cs Cesium
Cu Copper
F- Fluoride
Fe Iron
Fe(0) Zero-valent iron
FeCl42- Iron (II) tetrachloride anion
FeCO3 Iron (II) carbonate, ferrous carbonate
Fe3O4 Magnetite, iron (II,III) oxide
Fe2+ Aqueous form of the iron in the +2 oxidation state, ferrous anion
Fe3+ Aqueous form of the iron in the +3 oxidation state, ferric anion
Fe(II) Iron in the +2 oxidation state
Fe(III) Iron in the +3 oxidation state
Fe(OH)3 Ferric hydroxide
Fe(OH)2×(x-2)H2O Hydrated ferrous hydroxide
FeOOH Goethite, iron oxide hydroxide
FeS Iron (II) sulfide
H Hydrogen
H+ Hydrogen cation
H2 Hydrogen
HA Humic acid
HAal-LBr Aliphatic humic acid isolated from sediments collected from Lake Bradford, Florida, prepared by Florida State University
HAar-Gor Aromatic humic acid isolated from groundwaters near Gorleben, Germany, obtained from Professor J.-I. Kim, Institut für Radiochemie, München
HClO4 Perchloric acid
hmag. Hydromagnesite
HPO42- Hydrogenphosphate anion
HCO3- Bicarbonate anion, hydrogen carbonate anion
H2O Water
H2O2 Hydrogen peroxide
HOBr Hypobromous acid
HOCl Hypochlorous acid
H2PO4- Dihydrogen phosphate anion
H2S Hydrogen sulfide
K Potassium
K+ Potassium cation
K2MgCa2(SO4)4×2H2O Polyhalite
KNpO2CO3×2H2O Hydrated potassium neptunium (V) carbonate – (1:1:1) complex
K3NpO2(CO3)2×0.5H2O Hydrated potassium neptunium (V) carbonate – (3:1:2) complex
K2SO4 Potassium sulfate
K2U2O7 Potassium diuranate
mag. Magnesite
Mg Magnesium
Mg2+ Magnesium cation
MgCl2 Magnesium chloride
Mg3(OH)5Cl·4H2O Magnesium chloride hydroxide hydrate
MgCO3 Magnesite, magnesium carbonate
Mg5(CO3)4(OH)2×4H2O Hydromagnesite
Mg2(OH)3Cl×4H2O Magnesium chloride hydroxide hydrate, magnesium oxychloride
MgO Periclase, magnesium oxide
Mg(OH)2 Brucite, magnesium hydroxide
Mn Manganese
N2 Nitrogen
Na Sodium
Na+ Sodium cation
Na2SO4 Sodium sulfate
Na2S2O4 Sodium hydrosulfite
NaAm(CO3)2 Sodium americium (III) carbonate
NaCl Halite, sodium chloride
NaNpO2CO3×3.5H2O Hydrated sodium neptunium (V) carbonate – (1:1:1) complex
Na3NpO2(CO3)2 Sodium neptunium (V) carbonate – (3:1:2) complex
NaOH Sodium hydroxide
NaUO2O(OH)×H2O Clarkeite, sodium uranate
Na2U2O7×xH2O Sodium diuranate hydrate
Nd Neodymium
Nd(III) Neodymium in the +3 oxidation state
Nd(OH)3 Neodymium (III) hydroxide
Ni Nickel
Ni2+ Nickel (II) cation
Np Neptunium
Np(IV) Neptunium in the +4 oxidation state
Np(V) Neptunium in the +5 oxidation state
Np(VI) Neptunium in the +6 oxidation state
Np4+ Neptunium cation – Aqueous form of the neptunium at the +4 oxidation state
NpO2+ or Np(V)O2+ Neptunyl cation – Aqueous form of the neptunium at the +5 oxidation state
NpO22+ or Np(VI)O22+ Neptunyl cation – Aqueous form of the neptunium at the +6 oxidation state
NpO2CO3- Neptunium (V) carbonate ion – (1:1) complex
NpO2(CO3)23- Neptunium (V) carbonate ion – (1:2) complex
NpO2(CO3)35- Neptunium (V) carbonate ion – (1:3) complex
NpO2OH Neptunium (V) hydroxide
NpO2(OH)2- Neptunium (V) hydroxide ion – (1:2) complex
NO3- Nitrate anion
NS Adsorption site density (sites/nm2)
O Oxygen
O2 Molecular oxygen
OBr- Hypobromite anion
OCl- Hypochlorite anion
OH- Hydroxide anion
OH× Hydroxyl radical
Pb Lead
Pb(0) Zero-valent lead
Pb(II) Lead in the +2 oxidation state
Pb2+ Lead cation – Aqueous form of the lead at the +2 oxidation state
Pb4+ Lead cation – Aqueous form of the lead at the +4 oxidation state
PbCl2 Lead (II) chloride
PbCO3 Lead (II) carbonate
[Pb6O(OH)6]4+ Lead (II) polyoxyhydroxide cation
PbO Lead (II) oxide
PO43- Phosphate anion
(PbOH)2CO3 Lead (II) hydroxide carbonate
PbS Lead (II) sulfide
PbSO4 Lead (II) sulfate
Pu Plutonium
Pu(III) Plutonium in the +3 oxidation state
Pu(IV) Plutonium in the +4 oxidation state
Pu(V) Plutonium in the +5 oxidation state
Pu(VI) Plutonium in the +6 oxidation state
Pu(VII) Plutonium in the +7 oxidation state
Pu3+ Plutonium cation – Aqueous form of the plutonium at the +3 oxidation state
Pu4+ Plutonium cation – Aqueous form of the plutonium at the +4 oxidation state
Pu(CO3)+ Plutonium (III) carbonate ion – (1:1) complex
Pu(CO3)2- Plutonium (III) carbonate ion – (1:2) complex
Pu(CO3)33- Plutonium (III) carbonate ion – (1:3) complex
PuF22+ Plutonium (IV) fluoride cation
PuO2 Plutonium (IV) dioxide
PuO2+x Oxidized plutonium (IV) dioxide
PuO2CO3 Plutonium (VI) carbonate
PuO2CO3- Plutonium (V) carbonate ion – (1:1) complex
PuO2(CO3)23- Plutonium (V) carbonate ion – (1:2) complex
PuO2(CO3)22- Plutonium (VI) carbonate ion – (1:2) complex
PuO2(CO3)34- Plutonium (VI) carbonate ion – (1:3) complex
PuO2F+ Plutonium (VI) oxofluoride cation
PuO2+ or Pu(V)O2+ Plutonyl cation – Aqueous form of the plutonium at the +5 oxidation state
PuO22+ or Pu(VI)O22+ Plutonyl cation – Aqueous form of the plutonium at the +6 oxidation state
PuO3×xH2O Plutonium (VI) trioxide-hydrate
PuOH3+ Plutonium (IV) hydroxide cation – (1:1) complex
Pu(OH)22+ Plutonium (IV) hydroxide cation – (1:2) complex
Pu(OH)3+ Plutonium (IV) hydroxide cation – (1:3) complex
Pu(OH)4 Plutonium (IV) hydroxide
[Pu(H2O)m]n+ Hydrolysis complex of plutonium
[Pu(O)Pu(O)Pu(O)...]n Plutonium polymer
Ra Radium
S2- Sulfide anion
SO42- Sulfate anion
Sr Strontium
Tc(IV) Technetium in the +4 oxidation state
Th Thorium
Th(IV) Thorium in the +4 oxidation state
Th3+ Thorium cation – Aqueous form of the thorium at the +3 oxidation state
Th4+ Thorium cation – Aqueous form of the thorium at the +4 oxidation state
Th(CO3)56- Thorium (IV) pentacarbonyl ion complex
ThO2 Thorium dioxide
Th(OH)4 Thorium hydroxide
Th(OH)(CO3)45- Thorium (IV) hydroxide carbonate ion – (1:1:4) complex
Th(OH)2(CO3)22- Thorium (IV) hydroxide carbonate ion – (1:2:2) complex
Th(OH)3CO3- Thorium (IV) hydroxide carbonate ion – (1:3:1) complex
Th(OH)2SO4 Thorium (IV) hydroxide sulfate ion – (1:2:1) complex
Th(SO4)32- Thorium (IV) sulfate ion – (1:3) complex
Th(SO4)2 Thorium (IV) sulfate
U Uranium
U(III) Uranium in the +3 oxidation state
U(IV) Uranium in the +4 oxidation state
U(V) Uranium in the +5 oxidation state
U(VI) Uranium in the +6 oxidation state
U3+ Uranium cation – Aqueous form of the uranium at the +3 oxidation state
U4+ Uranium cation – Aqueous form of the uranium at the +4 oxidation state
U3O7 Triuranium heptaoxide
U4O9 Tetrauranium nonaoxide
UO2 Uraninite, uranium (IV) dioxide
UO22+ or U(VI)O22+ Uranyl cation – Aqueous form of the uranium at the +6 oxidation state
UO2CO3 Rutherfordine, uranium (VI) carbonate
UO2(CO3)22- Uranium (VI) carbonate ion – (1:2) complex
UO2(CO3)34- Uranium (VI) carbonate ion – (1:3) complex or triscarbonato complex
(UO2)3(CO3)66- Uranium (VI) carbonate ion – (3:6) complex
(UO2)2(CO3)(OH)3- Uranium (VI) carbonate hydroxide ion – (2:1:3) complex
(UO2)11(CO3)6(OH) 122- Uranium (VI) carbonate hydroxide ion – (11:6:12) complex
UO2OH+ Uranium (VI) hydroxide ion – (1:1) complex
(UO2)3(OH)5+ Uranium (VI) hydroxide ion – (3:5) complex
UO2(OH)3- Uranium (VI) hydroxide ion – (1:3) complex
UO2(OH)42- Uranium (VI) hydroxide ion – (1:4) complex
U(OH)3+ Uranium (IV) hydroxide ion – (1:3) complex
U(OH)4 Uranium (IV) hydroxide
UO2.xH2O Hydrous uranium (IV) dioxide
(UO2)(OH)2×xH2O or UO3×xH2O Schoepite, hydrated uranium trioxide
V Vanadium
ZrO2 Zirconium dioxide
Appendix SOTERM-2009 (Actinide Chemistry Source Term) is a summary of the U. S. Department of Energy’s (DOE’s) understanding of the Waste Isolation Pilot Plant (WIPP) chemical conditions, assumptions, and processes; the underlying actinide chemistry; and the resulting dissolved actinide concentrations that were calculated based on this repository chemistry. This appendix supplements Appendix PA-2009 in the 2009 Compliance Recertification Application (CRA-2009). The calculational results summarized here are based on the 2004 Performance Assessment (PA) Baseline Calculations (PABC) (Leigh et al. 2005), and hence on the various assumptions about chemical conditions in the repository that were included in the formulation of that baseline. WIPP-related geochemical experimental results obtained within and outside of the WIPP project since these calculations were performed are also summarized.
Actinide release from the WIPP is a critical performance measure for the WIPP as a transuranic (TRU) repository. There are a number of potential pathways for actinide release considered by the WIPP PA, and these are discussed in detail in Appendix PA-2009. Quantifying the impact of these releases contributes directly to assessing compliance with 40 CFR Part 191 (U.S. Environmental Protection Agency 1993).
In the undisturbed scenario for PA, actinide releases up the shafts or laterally through the marker beds are physically insignificant in all realizations and have no impact on compliance (Appendix PA-2009, Section PA-7.2). The self-sealing of the salt and the reducing anoxic environment in the repository provide the primary mechanisms for geologic isolation of the TRU waste in the undisturbed scenario. For the disturbed scenarios, actinide releases can occur primarily as a result of inadvertent human intrusions (i.e., boreholes drilled into or through the repository). For example, direct brine release (DBR) to the accessible environment may occur during a drilling intrusion, or actinides may be transported up a borehole to the Culebra Formation and then move laterally through the Culebra to the land withdrawal boundary (LWB). The potential for human intrusions makes it important to assess the range of possible repository conditions and associated dissolved actinide concentrations associated with the disturbed scenarios.
This appendix focuses on the actinide source term used to calculate actinide release from the WIPP for the DBR release and transport through the Salado and Culebra Formations. This actinide source term is the sum of the soluble and colloidal species in brine. Direct release of actinide particulates to the surface resulting from cuttings, cavings, and spallings is not considered part of the actinide source term because these particulate releases do not depend on the mobilized actinide concentrations in brine.
The relative importance of radioelements that significantly contribute to the actinide source term, and consequently impact the long-term performance of the WIPP, as established in the 2004 Compliance Recertification Application (CRA-2004) (U.S. Department of Energy 2004), Appendix SOTERM, and the CRA-2004 PABC is:
Pu » Am >> U > Th >> Np, Cm, and fission products. (SOTERM.1)
The TRU components for this list of radionuclides are the α-emitting isotopes of plutonium (Pu), americium (Am), neptunium (Np), and curium (Cm) with half-lives greater than 20 years. These TRU actinides make up the waste unit factor used to calculate the normalized release from the WIPP in U.S. Environmental Protection Agency (EPA) units, as required by Part 191. In SOTERM, the chemistry of thorium (Th) and uranium (U) is also discussed, since these actinides are present in the WIPP waste and their chemistry is analogous to the TRU components.
This appendix has the following overall organization:
· An overview of key near-field conditions and biogeochemical processes is presented in Section SOTERM-2.0.
· An updated literature review and summary of WIPP-relevant results for the key actinides is given in Section SOTERM-3.0.
· A summary of the WIPP actinide PA approach and assumptions, along with the calculated actinide solution concentrations, are provided in Section SOTERM-4.0.
· The PA implementation of the dissolved and colloidal components of the source term is described in Section SOTERM-5.0.
Each of these sections identifies important changes and/or new information since the CRA-2004 and the CRA-2004 PABC (Leigh et al. 2005).
The preemplacement and postemplacement near-field processes and conditions that could affect actinide concentrations in the WIPP are discussed in this section. An up-front summary of the current WIPP chemistry model assumptions is given in Table SOTERM-1, with a more detailed discussion of each assumption presented in the following sections. Emphasis is placed on how these processes and conditions in the repository could affect the concentrations of dissolved and colloidal actinide species in brine.
The ambient geochemical conditions are discussed in detail in the CRA-2004, Chapter 6.0. The Salado, which is the repository horizon, is predominantly pure halite (NaCl), with interbeds (marker beds) consisting mainly of anhydrite (CaSO4). The nearly pure halite contains accessory evaporite minerals such as anhydrite (CaSO4), gypsum (CaSO4×2H2O), polyhalite (K2MgCa2(SO4)4×2H2O), magnesite (MgCO3), and clays. Small quantities of intergranular (grain-boundary) brines and intragranular brines (fluid inclusions) are associated with the salt at the repository horizon. These brines are highly concentrated solutions (ionic strength up to 8 moles per liter [M]) of predominantly sodium (Na+), magnesium (Mg2+), potassium (K+), chloride (Cl-), and sulfate (SO42-), with smaller amounts of calcium (Ca2+), carbonate (CO32-), and borate (B(OH)4- and/or B4O72-). These brines have been in contact with the Salado evaporite minerals since their deposition (estimated to be 250 million years) and are saturated with respect to these minerals.
Underlying the Salado Formation is the Castile, composed of alternating units of interlaminated carbonate, anhydrite, and nearly pure halite. The Castile in the vicinity of the WIPP site is known to contain localized brine reservoirs with sufficient pressure to force brine to the surface if penetrated by a borehole. Castile brines are predominantly saturated NaCl solutions containing Ca2+ and SO42-, as well as small concentrations of other elements, and are about eight times more concentrated than seawater. Overlying the Salado in the vicinity of the WIPP site is the Culebra of the Rustler Formation, a fractured dolomite (CaMg(CO3)2) layer. It is significant because it is expected to be the most transmissive geologic pathway to the accessible environment. Culebra brines are generally more dilute than the Salado and Castile brines, and are predominantly NaCl with K+, Mg2+, Ca2+, SO42-, and CO32-. More detailed information on the distribution of Culebra brine salinity in the WIPP site and vicinity can be found in Appendix HYDRO-2009.
Repository conditions that could potentially affect actinide solubility are briefly summarized in this section. These include: repository pressure, repository temperature, water content and relative humidity, the minimum free volume for actinide release (effective porosity), and the extent of the disturbed rock zone (DRZ).
Table
SOTERM-1. Summary of Current
WIPP Chemistry Model Assumptions
(Leigh et al. 2005)
|
Repository Condition or Parameter |
CRA-2004/CRA-2004 PABC Assumptions |
SOTERM-2009 Section Containing References |
|
Ambient Geochemistry |
Predominantly halite of the Salado Formation, with anhydrite interbeds and inclusions. |
|
|
Temperature |
Ambient temperature is 28 oC (82 °F). An increase of up to 3 oC (5.4 °F) is possible as a result of the emplacement of TRU waste. |
|
|
Humidity |
~70 percent (%) relative humidity (RH) at the repository temperature. |
|
|
Water Content |
Host rock is groundwater-saturated with inclusions in the salt that range from 0.057% to 3% by mass. Repository is unsaturated for up to 1000 years (yr) depending on pressure and intrusion scenarios. |
SOTERM-2.2.3 |
|
Pressure |
A lithostatic pressure of about 15 megapascals (MPa) (148 atmospheres [atm]) at repository depth; a hydrostatic pressure of about 8 MPa (79.0 atm) at the bottom of an intrusion borehole at repository depth. |
|
|
Gas Phase |
Initially air/oxic at repository closure, but rapidly transitions to an anoxic atmosphere dominated by hydrogen with smaller amounts of methane and nitrogen. Trace amounts of carbon dioxide, hydrogen sulfide, and other microbially produced gases may be present. |
SOTERM-2.2.3 |
|
DRZ |
Upper bound of 12 meter (m) above the repository and 2 m below the repository horizon. |
|
|
Minimum Brine Volume for DBR |
The calculated minimum volume of brine from any source needed for DBR release is 10011 cubic meters (m3). |
|
|
WIPP Brine |
High-ionic-strength brine bracketed by Generic Weep Brine (GWB) and ERDA-6 brine formulations |
|
|
pH |
pH of about 9 and controlled by MgO, borate, and carbonate. |
|
|
MgO |
Engineered barrier for the WIPP that will sequester carbon dioxide (CO2) and control increases and decreases in pH by the precipitation of brucite, hydromagnesite, and magnesite. |
|
|
Microbial Effects |
Gas generation, primarily carbon dioxide and hydrogen sulfide, resulting from the biodegradation of cellulosic, plastic, and rubber (CPR) materials and creation of reducing conditions, including bioreduction of actinide elements from higher oxidation states. |
SOTERM-2.4.1 |
|
Corrosion |
Container steel and metals in WIPP waste will react to remove oxygen and produce hydrogen. |
|
|
Radiolysis |
Localized oxidizing effects possible near high-activity actinides, but overall radiolytic processes are overwhelmed by the in-room chemistry. |
The preexcavation lithostatic pressure (Stein 2005, CRA-2004 PABC, Section 4.1.1) in the WIPP at repository depth is about 15 MPa (148 atm). This pressure can be reestablished after repository closure due to salt creep and gas generation, but there are a number of PA vectors that predict pressure may not be fully restored even by the end of the 10,000-yr period of WIPP performance, and final pressures may range from 6 to 15 MPa (in the undisturbed scenario) and from 0.1 to 15 MPa (in the disturbed scenarios) considered in the CRA-2004 PABC. In this context, the pressure in the repository after closure cannot significantly exceed the far-field confining stress of about 15 MPa.
DBR can occur when the pressure in the repository at the time of a drilling intrusion exceeds 8 MPa and a sufficient amount of brine has already flowed into the repository (see related discussions in Section SOTERM-2.2.4 and Stein 2005). Eight MPa is the pressure exerted by a column of brine-saturated drilling fluid at the depth of the repository (Stoelzel and OBrien 1996). For repository pressures less than 8 MPa, no DBRs are assumed to occur because the fluid pressure in the repository cannot eject the drilling fluid from the borehole. There is also no DBR release until the brine volume exceeds the minimum brine volume (see Section SOTERM-2.2.4) needed to fill the effective porosity present in the compacted TRU waste.
When discussing the possible range of brine pressure to the source term, it is important to assess the possibility that the pressures experienced in the WIPP could impact actinide solubilities. In this context, the maximum pressure possible (~15 MPa) is well below pressures needed to affect the solution chemistry, and is not expected to have a significant effect on actinide solubilities or processes that lead to the association of actinides with colloidal particles. For these reasons, the effect of pressure on actinide solubility is not considered in the WIPP PA.
The ambient preemplacement temperature at the WIPP repository horizon was established to be 28 ºC (82 ºF) (Munson et al., 1987). The emplacement of TRU waste in the WIPP is expected to increase the ambient temperature by only a few degrees Celsius at most (Sanchez and Trellue 1996, Wang and Brush 1996a). For the purposes of PA, the temperature of the WIPP underground repository is assumed to be constant with time at 300 Kelvin (K) (27 ºC [80 ºF]) (Appendix PA-2009).
Actinide solubilities were calculated in WIPP PA using thermodynamic and laboratory data measured at 25 ºC [77 ºF]. The expected effect of the slightly elevated temperature in the WIPP on actinide concentrations is relatively small, especially when compared to other uncertainties inherent in the measurement and calculation of the actinide solubilities and colloidal concentrations. For this reason, the very small effect of temperature on actinide solubility was not considered in WIPP PA calculations.
A key argument for the WIPP as a TRU waste repository is that the self-sealing of the salt will limit the availability and transport of water into and through the repository, and correspondingly minimize the potential release of TRU from the repository. In all the undisturbed repository scenarios considered by PA, no actinide release from the WIPP is predicted (Leigh et al. 2005). There is, however, groundwater in the WIPP, even in undisturbed scenarios, that is potentially available to interact with the TRU waste. The salt surrounding waste is groundwater-saturated with both intergranular and intragranular water. The amount of water present as inclusions in the salt was used as a random variable in PA calculations (Leigh et al. 2005) with a range of 0.057 to 3 mass % based on what was measured in preexcavation salt (Skokan et al. 1987 and Powers et al. 1978). This brine can seep into the repository horizon and fill the excavated areas (TRU waste). Brine saturation of the repository is estimated to occur in less than 1000 years after repository closure.
The presence of some brine in the WIPP prior to brine saturation leads to an environment that will contain an atmosphere of up to about 70% RH, defined by the vapor pressure of saturated brine at the repository temperature. This water vapor pressure will be present, at least in part, until brine saturation occurs as a result of some human intrusions or brine seepage into the excavated area.
The presence of a humid environment in the WIPP prior to brine saturation may have a transitory effect on actinide solubilities. These transitory/temporary phases are not considered in WIPP PA because they will be rapidly overwhelmed by the in-room chemistry and higher reactivity of the waste components should brine inundation or saturation occur.
The minimum brine volume is the volume of brine needed for a DBR to occur during an intrusion scenario. There have been two calculational efforts to estimate this volume in the compacted TRU waste for the WIPP. Prior to the 1996 Compliance Certification Application (CCA) (U.S. Department of Energy 1996), Larson (1996) estimated this volume, calculated at 2000 years after repository closure with an assumption of no gas generation, to be 343 m3 per room and a minimum brine saturation of 0.75 for 116 equivalent rooms. This led to a repository-scale volume of 29,841 m3. Under these assumptions, this was the minimum brine volume needed for DBR.
Since this initial calculation, new information has led to a reassessment of this minimum volume (Stein 2005). The most important changes in this new calculation were: (1) it was based on the structural results used in the most current PA, (2) the time of the calculation was extended from 2000 to 10,000 years after repository closure, (3) a corrected waste-filled repository volume was used, and (4) the calculation was made to be more in line with the DBR conceptual model that requires a hydrostatic pressure of about 8 MPa. These changes led to a calculated per-room volume of 301.5 m3, a reduction in the minimum brine saturation value to 0.276, and 120.3 equivalent rooms. This led to an overall repository-scale volume of 10,011 m3.
The minimum repository brine volume has two important potential impacts on calculating actinide concentrations in the WIPP. The first is that the predicted inventory of some actinides, when fully dissolved in this brine volume, lead to concentrations that are below their predicted solubility, most importantly Np and Cm. In this context, they are assumed to be fully dissolved in the brine and may have an insignificant impact on the calculated actinide release in WIPP PA based on inventory arguments alone. The second impact is on the predicted concentration of key organic and inorganic complexants that coexist with the TRU species in WIPP waste. The maximum concentrations of acetate, citrate, and ethylenediaminetetraacetic acid (EDTA) (see Section SOTERM-2.3.6) are defined by their fully dissolved concentration in the minimum brine volume.
More detailed discussions of the DRZ can be found in Appendix PA-2009. The DRZ is a zone immediately surrounding the excavated repository that has been altered by the construction of the repository. In the Brine and Gas Flow (BRAGFLO) code, the Upper DRZ has a height of about 12 m (39 feet [ft]) and the Lower DRZ has a depth of about 2.2 m (7.2 ft) (Leigh et al. 2005, Figure 4-1). The creation of this DRZ disturbs the anhydrite layers and marker beds and alters the permeability and effective porosity of the rock around the excavated areas, providing enhanced pathways for the flow of gas and brine between the waste-filled rooms and the nearby interbeds.
The DRZ is important to the calculation of dissolved actinide concentrations because it potentially makes the minerals in the interbeds “available” for reaction with the TRU and emplaced waste components. The most important of these minerals is the calcium sulfate (anhydrite) that could function as a source of sulfate for processes in the repository subsequent to brine inundation. Currently, sulfate is assumed to be available from the DRZ into the waste area, which prolongs microbial sulfate reduction processes in the WIPP.
Brine present in the WIPP will react with emplaced TRU waste, waste components, and engineered barrier materials to establish the brine chemistry that will define actinide solubilities and colloid formation. In this context, the composition of the brine in the repository horizon will be defined by a combination of factors, including the initial composition of the in-flow brine; reactions that control pH; and the extent to which this brine is altered by equilibration with the waste components, emplaced container materials, and the waste-derived organic chelating agents that can dissolve in the brine. An overview of this repository chemistry is given in this section.
The composition of brine in and around the WIPP site prior to waste emplacement was established by sampling the groundwater and intergranular inclusions in the Salado and Castile (Popielak et al. 1983, Snider 2003a). Synthetic brines that simulate these compositions were developed and have been used for WIPP laboratory studies. The two simulated brines that best represent these repository-relevant, end-member brines are: (1) GWB, which simulates intergranular (grain-boundary) brines from the Salado at or near the stratigraphic horizon of the repository (Snider 2003a); and (2) ERDA-6, which simulates brine from the ERDA-6 well, typical of fluids in Castile brine reservoirs (Popielak et al. 1983). The concentrations of key inorganic species in these two brines, along with some brine properties, are listed in Table SOTERM-2.
Table SOTERM-2. Compositions of GWB and ERDA-6 Prior To and After Equilibration with MgO (Brush et al. 2006)
|
Ion or Propertya |
GWBb |
GWB |
ERDA-6d Before Reaction with MgO, Halite, and Anhydrite |
ERDA-6 |
|
B(OH)x3-x (see Footnote e) |
158 mM |
166 mM |
63 mM |
62.4 mM |
|
Na+ |
3.53 M |
4.35 M |
4.87 M |
5.24 M |
|
Mg2+ |
1.02 M |
0.578 M |
19 mM |
157 mM |
|
K+ |
0.467 M |
0.490 M |
97 mM |
96.1 mM |
|
Ca2+ |
14 mM |
8.95 mM |
12 mM |
10.7 mM |
|
SO42- |
177 mM |
228 mM |
170 mM |
179 mM |
|
Cl- |
5.86 M |
5.38 M |
4.8 M |
5.24 M |
|
Br- |
26.6 mM |
27.8 mM |
11 mM |
10.9 mM |
|
Total Inorganic C (as HCO3-) |
Not reported |
0.35 mM |
16 mM |
0.428 mM |
|
pH |
Not reported |
8.69 |
6.17 |
8.94 |
|
Relative Density |
1.2 |
1.23 |
1.22 |
1.22 |
|
Ionic Strength (m) |
7.56 |
7.66 |
6.05 |
6.80 |
|
a Ions listed represent the total of all species with this ion. b From Snider (2003a) c From Brush et al. (2006) d From Popielak et al. (1983) e Boron species will be present in brine as boric acid, hydroxy polynuclear forms (B3O3(OH)4-, and/or borate forms (e.g., B4O72-) |
||||
At the time of the CCA, Brine A (Molecke 1983) and Salado Primary Constituents (SPC) Brine, a version of Brine A from which trace elements had been removed, were used to simulate Salado brines for laboratory and modeling studies. Since the CCA, however, GWB has been shown to be more representative of intergranular Salado brines than either Brine A or SPC Brine (Brush and Xiong 2003a, Snider 2003a). This brine formulation is currently used to represent Salado brines in PA. In particular, the magnesium concentration of GWB (1.0 M) simulates the average concentration of this element in Salado brines more closely than Brine A (1.44 M).
The reaction with MgO, based on the modeling calculations performed, leads to some potentially significant changes in the composition of the brine (see Table SOTERM-2). The most important of these changes for GWB brine is the lowering of the magnesium concentration from 1.02 to 0.578 M, a decrease in calcium concentration from 14 to 8.95 mM, and a pH of 8.69. For ERDA-6, there is a significant increase in the magnesium concentration from 19 to 157 mM, a decrease in total inorganic carbon from 16 to 0.428 mM, and an increase of the pH to 8.94 from 6.17. The pH associated with these MgO-reacted brines established the range of expected pH values in the WIPP for the calculation of actinide solubilities, and the composition of these reacted brines were used in PA to calculate actinide solubility in brine (Brush 2005).
Salado brine will enter the repository after closure, and can be supplemented by Castile brine in some human intrusion scenarios. It is also possible that groundwater from the Rustler and Dewey Lake Formation could flow down the borehole into the repository, mix with the waste, and then be forced back up a borehole. The majority of WIPP-specific solubility studies since the CRA-2004 were performed using GWB or ERDA-6 brines, since these brines bracket the expected range in brine composition. Including brine mixing in PA has been considered and rejected because using the end member brines (i.e., GWB or ERDA-6 brines) brackets the median values and uncertainties for the solubility calculations.
In addition to using these end-member brines in PA, other simplifying assumptions were also made:
1. Any brine present in the repository is well mixed with waste.
2. Equilibria with halite and anhydrite, the most abundant Salado minerals at or near the stratigraphic horizon of the repository, are rapidly established.
3. Oxidation-reduction (redox) equilibria with waste materials were not assumed.
4. Brine compositions attained after equilibration of GWB or ERDA-6 with the MgO engineered barrier exist for the entire 10,000-year regulatory period.
Brine composition is important to the calculation of actinide concentrations. The inorganic complexants, ionic strength, and pH are direct inputs needed to calculate actinide solubilities for a given brine composition. These species and properties are also important in defining the potential for colloid formation in the WIPP.
The brine pH is a very critical parameter in defining the solubility of actinides under conditions where brine-mediated releases (DBR and transport through the Culebra) would be important in the WIPP. The brine pH is established by a number of highly coupled processes that will occur when the emplaced WIPP waste is inundated with brine. The most important of these are the potential buffering capacity of the brine coming into the WIPP, the reactions of this brine with emplaced waste components (most notably reduced metals and MgO), and microbial processes. The reactions of the emplaced MgO barrier material are expected to sufficiently control and define the pH when the repository is saturated with brine.
The range of brine composition that is likely to be present in the WIPP repository was discussed in Section SOTERM-2.3.1 (see also Table SOTERM-2). These brines have an intrinsic buffering capacity that is highest at pH 8.5-9. ERDA-6 brine, although it has an ambient pH of 6.2, contains a number of constituents that, in the pH range of 8-10, add buffer capacity to the reacted brine: Carbonate/bicarbonate (16 mM), borate (63 mM), and divalent cations that tend to react with hydroxide or carbonate to influence pH (Ca2+ at 12 mM, and Mg2+ at 19 mM). The pKa for boric acid and dissolved carbonate/bicarbonate species are 9.0 and 9.67, respectively, which explains the tendency of this brine to maintain the pH in the range of 8-10. Operationally, the simulated ERDA-6 brines prepared in the laboratory have relatively high buffering capacity, and significant changes in brine concentrations and pH are not routinely observed once the pH is experimentally defined (Borkowski et al. 2008, Lucchini et al. 2009). An operational pH range for ERDA-6 has been defined as having an upper limit of pH ~10, which is the pH at which a cloud point (indicating Mg precipitation) is observed. The preexcavation ambient ERDA-6-like brine will naturally add to the buffering capacity of WIPP brine due to its acid-base components and will establish a relatively high buffer capacity at the mildly alkaline conditions expected in the WIPP.
The expected pH in the WIPP in the event of brine saturation, however, will be defined by the reaction of the Castile ERDA-6-like brine with the waste components and barrier material. This was evaluated as part of the documentation for the CRA-2004 PABC (Leigh et al. 2005, Brush et al. 2006, and Table SOTERM-2).
Under these repository-relevant conditions, the expected pH, when little or no carbonate is present, is 8.69 in GWB brine and 8.94 for ERDA-6 brine. In both cases, this pH is established/buffered by the brucite dissolution reaction. The presence of microbial activity will potentially contribute significant amounts of carbon dioxide and leads to a model-predicted pH of 8.69 and 9.02 for GWB and ERDA-6 brine, respectively.
The key role MgO has in the buffering of pH at 8 to 9 under WIPP-relevant conditions is the basis of current WIPP actinide solubility calculations. In the absence of significant amounts of CO2, the following carbonation reaction will buffer the fugacity of carbon dioxide (fCO2) at a value of 10-5.48 atm in GWB and 10-6.15 atm in ERDA-6:
Mg(OH)2 + Ca2+ + CO2(aq or g) D CaCO3 + Mg2+ + H2O(aq or g) (SOTERM.2)
Under these conditions, the following brucite dissolution/precipitation reaction will buffer pH in the WIPP at a value of 8.69 in GWB and 8.94 in ERDA-6 (Brush et al. 2006).
Mg(OH)2 D Mg2+ + 2OH- (SOTERM.3)
The potential for significant CO2 formation as the result of microbial activity changes the mechanism by which pH is buffered, but causes a relatively small change in the calculated pH for ERDA-6-like brines. Microbial consumption of CPR materials could produce significant quantities of CO2, which could in turn acidify any brine present in the repository and increase the solubility of the actinides relative to that predicted for near-neutral and mildly basic conditions. Under these conditions, both laboratory and modeling studies predict that the following carbonation reaction:
5Mg(OH)2 + 4CO2(aq or g) D Mg5(CO3)4(OH)2×4H2O (SOTERM.4)
will buffer fCO2 at a value of 10-5.50 atm in both GWB and ERDA-6. In this reaction, Mg(OH)2 is the brucite, which is the main hydration product of the periclase (MgO) expected in the WIPP; Mg5(CO3)4(OH)2×4H2O is the form of the hydromagnesite expected in the repository. Consideration of the possibility of high CO2 levels leads to a calculated pH of 8.69 and 9.02 for GWB and ERDA-6 brine, respectively. This is a relatively small change in the predicted pH with no change predicted in GWB brine and only a 0.08 pH shift in ERDA-6 brine. These values of fCO2 and pH were used in the actinide speciation and solubility calculations for all CRA-2004 PABC vectors (Brush 2005).
Experiments that are relevant to the chemistry and pH buffering capacity of MgO, but not reflected in the CRA-2004 PABC, were performed by investigators at Karlsruhe (Schuessler et al. 2001 and Altmaier et al. 2003). This research was done to support the development of the German salt-based repository where MgO, calcium oxide (CaO), and clays are being evaluated as potential backfill material. Equilibration experiments with 2.67 and 5.15 molal magnesium chloride with excess magnesium hydroxide present were conducted for durations of over 400 days and show the establishment of a stable magnesium solution concentration with a pH of 8.7 to 8.8, which is in excellent agreement with current WIPP model predictions (see Figure SOTERM-1). This equilibration was also modeled using the Pitzer formulation in the software package for geochemical modeling of aqueous systems (EQ3/6), and excellent agreement was obtained. In this study, a change in magnesium (Mg) concentration was not noted during the equilibration with MgO, even though cement was dissolved in brine.
Based on Figure SOTERM-1, the dissolution of MgO in brine, when high chloride concentrations are present (> 2 m), demonstrates the self-buffering property of the brine-MgO system. The pH does not increase when MgO is dissolved. Moreover, with time, the system counteracts the potential decrease in pH that could result from solid phase transformations. To illustrate this better, the dissolution of 1 mole of MgO introduces 1 mol of Mg2+ and 2 moles of OH- to the brine, which should increase the pH. To counter this and maintain pH, the magnesium chloride hydroxide hydrate (Mg2(OH)3Cl×4H2O) phase precipitates to reduce the pH. For the data shown in Figure SOTERM-1, the pH is reduced slowly as a consequence of equilibration with MgO. In this context, more hydroxide ions are precipitated than are introduced to the brine during the MgO dissolution step. There will also be more magnesium precipitated from the brine, resulting in a lower magnesium concentration in the brine.
There are no new WIPP-specific results to report
that explicitly address the MgO buffering of WIPP brine since the
CRA-2004. Some WIPP-specific experiments using simulated
GWB and ERDA-6 brine, however, indirectly provide some
information on this subject (Xiong and Lord 2008, Lucchini et al. 2009, Borkowski et al. 2008, Reed et al. 2009). A considerable number of the solubility
experiments were performed in the pH range of 8-10 (below the
cloud point of either ERDA-6 or GWB brine) and reflect strong
buffering with no pH drift over the greater than 2-year
duration of the experiments. Additionally, no significant
Mg precipitation was noted in this pH range. The brines in
these solubility studies were not equilibrated with MgO, but in
some cases had excess iron in the system. Several
experiments were performed outside of this pH range; in the
presence of high carbonate (10 mM), a slow, downward pH drift was
observed that was as much as 2 pH units over the duration of the
experiments, even through preequilibration at the desired higher
experimental pH was initially performed. In Xiong and Lord
(2008), where the MgO and brucite reaction paths in GWB, ERDA-6
brine, and simplified brines were investigated, the equilibrium
pH values measured were pH about 9 and were established by the
reaction/dissolution of the MgO or Mg(OH)2.
Slightly higher pH was noted (up to pH 9.7) in some simplified
brines when no carbonate or other brine components were
present. All of these data, although indirect, suggest that
the MgO controls the pH to a pH = 9 ±
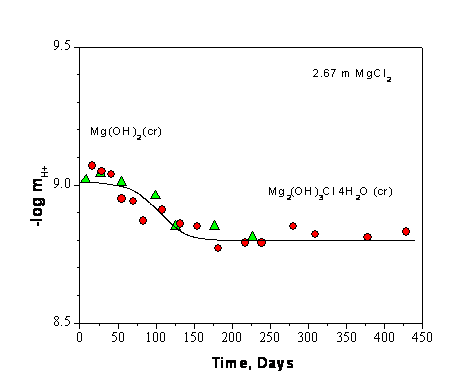
Figure SOTERM-1. Molal H+ Concentration Measured as a Function of Time During the Solubility Experiments in 2.67 and 5.15 m MgCl2 Solution. The Filled Circles and Triangles Show the Two Experiment Runs (Based on Data in Altmaier et al. 2003, Figure 3).
1. In this context, it is predicted that brine pH will remain in the range of 8-10 under a wide range of expected conditions. These experimental observations are also consistent with the experimental results and model predictions reported by Altmaier et al. (2003).
MgO is the bulk, granular material emplaced in the WIPP as an engineered barrier. The MgO currently being placed in the WIPP contains 96 ± 2 mol % reactive constituents (i.e., periclase and lime) (Deng et al. 2006). The amount of MgO emplaced in the WIPP is currently calculated based on the estimated CPR content with an excess factor of 1.2, and it is estimated that in excess of 75,000 metric tons will be emplaced in the WIPP by the time of repository closure.
The chemistry of MgO is critical to the overall performance of the WIPP and is discussed in detail in Appendix MgO-2009 and in Xiong and Lord (2008). The MgO, as an engineered barrier in the WIPP repository design, has two important functions that directly support the PA calculation of actinide concentrations in brine. These are:
1. Sequester the excess CO2 produced by the microbial consumption of CPR material and establish/maintain a low fCO2 in the repository. This is currently estimated to be 10-5.5 atm for GWB and ERDA-6 brine.
2. Establish and buffer the brine pH by maintaining a magnesium solution concentration that reacts with CO2 and hydroxide (see reaction SOTERM.2 and SOTERM.3) to buffer the pH at about 9. This was part of the pH discussion in Section SOTERM-2.3.2. This buffering removes uncertainty from the actinide concentration calculations.
Initially, MgO will undergo hydration to generate brucite (Mg(OH)2). In time, brucite will react further to form magnesium chloride hydroxide hydrate (Mg3(OH)5Cl·4H2O) in Salado brine (Appendix MgO-2009, Section MgO-4.1). These phases combine to control the concentration of magnesium in high-magnesium brine (for example, GWB). The existence of magnesium as an aqueous cation in equilibrium with excess magnesium minerals helps to establish the solution pH.
For the reaction of MgO with GWB brine, PA uses a magnesium concentration of ~0.6 M (see Table SOTERM-2), which is supported by experimental results showing a magnesium concentration ~0.7 M (Snider 2003b). This reaction was also investigated by Altmaier et al. (2003) and Harvie, Møller, Weare (1984). Snider noted that the rate of MgO hydration is most likely linked to mineral phase changes between hydrated magnesium oxychloride and brucite. The existence of the hydrated magnesium oxychloride phase was inferred from scanning electron microscope (SEM) images, coupled with an energy dispersive x-ray spectroscopy system (EDS), to identify Mg–Cl phases. The Altmaier and Harvie studies showed that the hydration reaction was a solid-phase transformation between brucite and hydrated magnesium oxychloride that depends not on magnesium concentration, but on chloride concentration, with an invariant point predicted at 1.8 m MgCl concentration and a –log mH+ = 8.95.
The most important role of the MgO engineered barrier is to sequester carbon dioxide to maintain a low fCO2 in the repository. Microbial consumption of CPR materials could produce significant quantities of CO2. Under these conditions, brucite and magnesium chloride hydroxide hydrate will react with the CO2 generated. Both laboratory and modeling studies predict that the following carbonation reaction will buffer fCO2 at a value of 10-5.50 atm in both GWB and ERDA-6:
5Mg(OH)2 + 4CO2(aq or g) D Mg5(CO3)4(OH)2×4H2O (SOTERM.5)
This reaction effectively removes excess CO2 from the repository and bicarbonate/carbonate from the brine. The initial product of MgO carbonation reaction is Mg5(CO3)4(OH)2·4H 2O. This is converted into MgCO3, which is the expected stable mineral form of magnesium carbonate in the WIPP, according to Reaction (SOTERM.6).
Mg5(CO3)4(OH)2×4H2O + CO2(aq or g) + 10 H2O D 5MgCO3∙H2O (SOTERM.6)
Reaction (SOTERM.6) is slow and it is estimated that hundreds to thousands of years (Appendix MgO-2009) are needed for the conversion of hydromagnesite to magnesite. Consumption of CO2 will prevent the brine acidification, and magnesium carbonate precipitation will maintain low carbonate concentration in the WIPP brine to avoid the formation of highly soluble actinide species with carbonate complexes. Although MgO will consume essentially all CO2, residual quantities in equilibrium with magnesite under the WIPP conditions will persist in the aqueous and gaseous phases.
The importance of magnesium chemistry, and correspondingly the chemistry associated with the emplaced MgO on the calculation of actinide concentrations in brine is clear. MgO sequesters CO2 and minimizes the buildup of carbonate in brine. At the expected pH, carbonate forms strong complexes with the An(III), An(IV), and An(VI) oxidation states. An increased carbonate concentration in brine would significantly increase actinide solubilities. Additionally, MgO helps establish the pH in brine. The removal of CO2 prevents a decrease in the pH that could also significantly increase actinide solubility. An additional beneficial effect of MgO is to maintain a solution concentration of Mg2+ that will precipitate as brucite to keep the pH in the 8-10 range. The presence of MgO leads to a more predictable chemistry that lowers the uncertainty when calculating actinide concentrations in WIPP brine (see Borkowski et al. 2008 for data on An(III) and Altmaier et al. 2005 for data on An(IV)).
The WIPP repository will contain a large quantity of reduced iron due to the use of iron-based containers for much of the emplaced TRU waste. Currently, it is estimated that the WIPP will contain upwards of 51,000 metric tons of iron (U.S. Department of Energy 2006) when all the waste is emplaced. The presence of this reduced metal will have an important role in the establishment of reducing conditions in the WIPP by removing oxygen. Reduced iron species (aqueous Fe(II) and Fe(0, II)-valent minerals) are important because they will reduce higher-valent actinides in the WIPP, leading to lower actinide solubilities (Reed et al. 2009, Reed et al. 2006).
It is expected that oxic corrosion of steels and aerobic microbial consumption of CPR materials will quickly consume the limited amount of oxygen (O2) trapped within the repository at the time of closure. After O2 is consumed, anoxic corrosion of metals will occur (Brush 1990, Brush 1995, Wang and Brush 1996a). In all of the vectors for the 2004 PA, the EPA’s CCA 1997 Performance Assessment Verification Test (PAVT), the CCA PA, and the CRA-2004 PABC, there were significant amounts of uncorroded steels and other Fe-base alloys in the repository throughout the 10,000-yr regulatory period. WIPP-specific experiments (Telander and Westerman 1993 and 1997) showed that steels and other Fe-based alloys will corrode by the following reactions:
Fe + (x)H2O D Fe(OH)2×(x-2)H2O + H2; (SOTERM.7)
3Fe + 4H2O D Fe3O4 + 4H2; (SOTERM.8)
Fe + H2O + CO2 D FeCO3 + H2; and (SOTERM.9)
Fe + H2S D FeS + H2. (SOTERM.10)
In reducing environments, reduced iron phases (Fe(II) oxides and zero valent iron) and aqueous ferrous iron will be present. These are all reducing agents towards key actinide species (see Table SOTERM-3) and will help establish the predominance of lower-valent actinides in the WIPP. The concentration of ferrous iron could be relatively high in the WIPP brine, although its solubility has not yet been explicitly determined. There are also many potential reactions that could control and/or define the iron chemistry. The expectation is that ferrous hydroxide will control the solubility of iron, leading to a predicted solubility in the range of 10-6 M to 10-4 M for pH between 8.5 and 10.5 (Refait and Génin 1994).
Table SOTERM-3. Redox Half-Reaction Potentials for Key Fe, Pb, Pu, and U Reactions at 25 oC and I<1 (Morss, Edelstein, and Fuger 2006, Chapter 23)
|
Metal Species Reduced |
Eo (Acidic) in V |
Eo at pH = 8 in V |
|
Pb4+ → Pb2+ |
1.69 |
2.47 |
|
PuO2+ → Pu4+ |
1.170 |
0.70 |
|
PuO22+ → PuO2+ |
0.916 |
0.60 |
|
Fe(OH)3(s) →Fe2+ |
Not Applicable |
0.1 |
|
FeOOH (s)→FeCO3(s) |
Not Applicable |
-0.05 |
|
UO22+ → U4+ |
0.338 |
-0.07 |
|
Pu4+ → Pu3+ |
0.982 |
-0.39 |
|
Pb2+ → Pb |
-0.1251 |
-0.54 |
|
Fe3+→ Fe2+ |
0.77 |
-0.86 |
|
Fe(II)(OH)2 → Fe(0) |
-0.44 |
-0.89 |
|
U4+ → U3+ |
-0.607 |
-1.95 |
Three important reactions of iron are considered for the WIPP PA. The first is the reaction of metallic iron with carbon dioxide to form strongly insoluble ferrous carbonate. The solubility product of this salt is log K = -10.8 at I = 0 (National Institute of Standards and Technology [NIST] 2004), and it is much smaller than magnesium carbonate. This suggests that the presence of iron will likely remove CO2 from the repository more effectively than MgO due to its lower solubility product. This reaction is not included in the WIPP PA because the CO2 reacts with MgO before the iron.
The second is the reaction of iron and ferrous ions with the hydrogen sulfide that could be generated in the repository by sulfate-reducing microbes. This will lead to a very insoluble ferrous sulfide precipitate with a solubility product of log Ks = -17.2 (NIST 2004). This helps remove sulfide, which can complex actinides, from brine. This reaction is assumed to occur instantaneously in the PA.
Finally, iron species form strong complexes with organic ligands. The strongest of these complexes is EDTA. The net effect is that dissolved iron species will compete with actinides for organic ligands, and in many cases out-compete the actinides to counteract the potential enhancement of actinide solubility that would otherwise occur. This reaction is not currently included in the PA.
The chemistry of iron will have a pronounced effect on WIPP-relevant actinide chemistry in many ways. The linkages of iron chemistry to the redox chemistry are well-established in the literature (Farrell et al. 1999, Fredrickson et al. 2000, Qui et al. 2001, Nakata et al. 2004, and Behrends and Van Cappellen 2005). Iron will establish reducing conditions conducive to the overall reduction of higher-valent actinide species and precipitate an iron sulfide phase that removes sulfide from solution. Additionally, iron species could sequester carbon dioxide and compete with actinides for organic and inorganic complexants, although there is no explicit credit taken for this in the WIPP PA.
Lead is present in the repository in the metallic form as part of the waste. The reactivity of zero-valent lead is greatly mitigated by the formation of a thin, coherent, protective oxide, oxycarbonate, chloride, or sulfate protective layer. Metallic lead also reacts slowly with water at room temperature and undergoes corrosion to form oxides and oxyhydroxides. Under slightly alkaline conditions, the hydrolysis of lead leads to formation of a poly-oxyhydroxide cation, [Pb6O(OH)6]4+. The following reactions are possible under WIPP-relevant conditions:
2Pb + O2 D 2PbO (SOTERM.11)
2PbO + H2O + CO2 D (PbOH)2CO3 (SOTERM.12)
Pb + H2O + CO2 D PbCO3 + H2 (SOTERM.13)
Pb + H2S D PbS + H2 (SOTERM.14)
Pb2++ 2Cl- D PbCl2 (SOTERM.15)
Pb2++ SO42- D PbSO4 (SOTERM.16)
5Pb2++ PbO + 6OH- D [Pb6O(OH)6]4+ (SOTERM.17)
The solubility of lead in WIPP brine is expected to be low, due in part to the passivation process, but also because of insoluble solids formation. Strong oxidants, e.g., radiolysis products, may locally enhance the dissolution of lead, but alkaline brine, which contains chlorides and carbonate/bicarbonate species, will overwhelm radiolytic effects to maintain a low concentration of lead in the brine. In solution, lead will exist as Pb2+ species that are redox-active toward high-valent actinides (see Table SOTERM-3) and will help establish and maintain reducing conditions in the brine.
Lead, as was the case with iron, can influence the redox chemistry (see Table SOTERM-3) and precipitate carbonate and sulfide from the WIPP brine. This leads to a redox chemistry that will help maintain reducing conditions and effectively lower carbonate concentration. Both of these will potentially lower actinide solubility in the WIPP. These impacts are not considered in the WIPP PA.
Organic chelating agents are used in the processing and cleanup/decontamination of actinides throughout the DOE complex. For this reason, they are often present as cocontaminants with the TRU component in the WIPP waste. Some of these chelating agents strongly complex actinides and could have a significant effect on their solubility in brine. In this context, four organic chelating agents–oxalate, acetate, citrate, and EDTA–are tracked as part of the WIPP inventory process, and the potential effects of these complexants on the calculated actinide solubilities are evaluated as part of the WIPP PA (Leigh et al. 2005, Brush and Xiong 2005a).
The potential concentrations of the key organic ligands in the WIPP were calculated a number of times (Brush and Xiong 2003b, Leigh, Trone, and Fox 2005) and are based on the inventory provided by Crawford and Leigh (2003). The potential concentrations of these organics used in the CRA-2004 PABC were calculated by Brush and Xiong (2005a) and are based on the best understanding of the WIPP inventory data available at that time. These concentrations are summarized in Table SOTERM-4, where the potential maximum organic concentration in the WIPP is defined as the inventory of the organic ligand divided by the minimum free volume of brine needed for brine release (see Section SOTERM-2.2.4).
Dissolved metals will compete with the actinides to form organic complexes. As the metals in the repository corrode, additional transition metal ions will dissolve into the brine. These ionic species include iron (Fe) and lead (Pb). Other steel constituents, such as nickel (Ni), chromium (Cr), vanadium (V), and manganese (Mn), may also be present. Additionally, divalent cations in the brine, most importantly Mg2+ and Ca2+, will also form complexes with these chelating agents and compete with the actinide species. The stability constants for Mg2+, Ca2+, Fe2+, Pb2+, and Ni2+ and deprotonation constants for the organic acids are shown in Table SOTERM-5 (National Institute of Standards and Technology 2004). These formation constants, in many respects, follow the same trends as the actinide species and, when present in high enough concentrations, will compete with the actinide to form complexes and effectively lower the effect of organic complexation on actinide solubility. However, this is not included in the PA.
Table SOTERM-4. Concentrations of Organic Ligands in WIPP Brine Calculated for Use in the CRA-2004 PABC (Brush and Xiong 2005a)
|
Organic Ligand |
Compound |
Inventory Amount |
Molecular |
Potential Concentrationc |
Total Potential
Concentrationc |
|
Acetate |
Acetic acid Sodium acetate |
1.42 ´ 105 8.51 ´ 106 |
60.05 82.03 |
2.36 ´ 10-4 1.04 ´ 10-2 |
1.06 × 10-2 |
|
Oxalate |
Oxalic acid Sodium oxalate |
1.38 ´ 107 3.39 ´ 107 |
90.03 112.0 |
1.53 ´ 10-2 3.02 ´ 10-2 |
4.55 × 10-2b |
|
Citrate |
Citric acid Sodium citrate |
1.19 ´ 106 4.00 ´ 105 |
192.1 214.1 |
6.19 ´ 10-4 1.87 ´ 10-4 |
8.06 × 10-4 |
|
EDTA |
Sodium salt |
2.56 ´ 104 |
314.2 |
8.14 ´ 10-6 |
8.14 × 10-6 |
|
a Molecular weight was calculated for monosodium salts to be conservative. b Inventory, in moles, of the organic chelating agent divided by 10,011 m3 c Concentration of oxalate will be limited by solubility, not inventory, in ERDA-6-like brine |
|||||
Table SOTERM-5. Apparent Stability Constants for Organic Ligands with Selected Metals (National Institute of Standards and Technology 2004)
|
Organic Ligand |
pKa |
Metal |
Ionic Strength (m) |
log10 β |
|
EDTA |
k1 8.86-9.05 k2 6.10-7.02 k3 2.79-2.54 k4 2.05-2.20 |
Fe2+ Ni2+ Pb2+ Mg2+ Ca2+ |
0.1 0.1 0.1 1 1 |
14.3 18.4 18 8.61 9.68 |
|
Citrate |
k1 5.58-5.30 k2 4.25-4.38 k3 2.85-3.06 |
Fe2+ Ni2+ Pb2+ Mg2+ Ca2+ |
0.1 0.1 1.0 0.1 0.1 |
4.4 5.18 4.44 3.43 3.48 |
|
Oxalate |
k1 3.74-4.23 k2 1.15-1.43 |
Fe2+ Ni2+ Pb2+ Mg2+ Ca2+ |
1.0 0.1 1.0 0.1 0.1 |
3.05 4.16 4.20 2.75 2.46 |
|
Acetate |
k1 4.52-4.99 |
Fe2+ Ni2+ Pb2+ Mg2+ Ca2+ |
3.0 0.1 0.1 0.1 0.1 |
0.54 0.88 2.15 0.51 0.55 |
There are two final, but important, observations about the organic chelating agents present in the WIPP. First, they are expected to have very different tendencies toward biodegradation, based on extensive experience with soil bacteria in the literature (Banaszak, Rittmann, and Reed 1999). Microbial activity, based on many general observations with soil bacteria, will likely readily degrade citrate, oxalate, and acetate to very low (submicromolar) steady-state concentrations. This important degradation pathway is not as certain for EDTA, which tends to resist biodegradation in most groundwaters. These degradation pathways have, however, not been demonstrated for the halophiles typically present in the WIPP, and it is currently assumed in the WIPP PA that no degradation pathways for these organic complexants, microbiological or chemical, exist.
The second important observation is that these chelating agents, under WIPP-relevant conditions, are expected to help establish reducing conditions in the WIPP because they tend to reduce higher-valent actinides. This has been demonstrated in WIPP brine for Np(V) and Pu(V/VI), but was not observed for U(VI) (Reed et al. 1998). These chelating agents also tend to oxidize III actinides to IV, which would have a beneficial effect on actinide solubility in the WIPP because the actinides in the IV oxidation state are approximately 10 times less soluble than actinides in the III oxidation state. These potentially beneficial effects of organic chelating agents on actinide speciation are also currently not included in the WIPP PA.
The WIPP waste contains a relatively high amount of organic material, since much of the waste is residue from laboratory operations where CPR materials were widely used. Current estimates project over 10,000 metric tons of plastic and cellulosic materials with a much lower amount of rubber material in the WIPP. This organic material is important from the perspective of repository performance in that it provides an organic “feedstock” for microbial activity that could lead to gas generation (carbon dioxide, hydrogen, hydrogen sulfide, and possibly methane), as well as degradation products that can complex actinides or form pseudocolloids. CPR degradation is represented in the PA to evaluate these potential impacts on the actinide concentrations and release.
There are three important postemplacement processes that take place in the WIPP after repository closure. These are metal corrosion, microbiological effects, and radiolysis. Metal corrosion was already discussed as part of the iron chemistry section (Section SOTERM-2.3.4). Microbiological effects and radiolysis are briefly discussed in this section.
Microbiogical processes can have a significant effect on many aspects of subsurface chemical and geochemical processes. This, particularly as it relates to contaminant transport and remediation, has been well established for soil bacteria in low-ionic-strength and near-surface groundwaters (Banaszak, Rittmann, and Reed 1998). In the WIPP, as a result of the high-ionic-strength brines present, halophiles (rather than soil bacteria) will predominate. What is understood about halophiles under WIPP-relevant conditions was established through a series of long-term studies conducted as part of the Actinide Source Term Program (ASTP) project by researchers at Brookhaven (Brush 1990; Francis and Gillow 1994; Brush 1995; Wang and Brush 1996a). The important and potential effects of microbial activity on the WIPP PA are also discussed extensively (Leigh et al. 2005, U.S. Environmental Protection Agency 2006).
In the WIPP repository, many of the co-contaminants present (e.g., sulfates, phosphates, organics, nitrate) are important nutrients that drive microbial activity and, in part, select the primary degradation and growth pathways taken. There are WIPP-specific data which demonstrate that microbial processes can occur under humid and saturated conditions in the laboratory (Francis 1998). In the CRA-2004 PABC, a longer-term rate for microbial gas generation was implemented based on new laboratory data obtained by the project (Leigh et al. 2005; Nemer and Stein 2005; Nemer, Stein, and Zelinski 2005).
Under repository-relevant conditions, there are primarily two important potential effects on repository performance linked to the presence of microbial activity. The first is gas generation due to the biodegradation of the organics present as WIPP waste (see Section SOTERM-2.3.6 and SOTERM-2.3.7). This is currently addressed by the WIPP PA (Nemer and Stein 2005; Nemer, Stein, and Zelinski 2005). The second is the growing recognition of the linkages between microbial activity and actinide speciation under microbiologically active anaerobic and reducing conditions. This is not currently addressed in the WIPP PA, but adds an additional argument for the sustained predominance of lower-valent actinides under WIPP-relevant conditions (III and IV oxidation states).
Microorganisms utilize organic compounds as the carbon source for their growth. This biodegradation also provides energy to the organism because the organic compound also functions as an electron donor that, when coupled with inorganic electron acceptors (oxidized metals, sulfate, nitrate, and or oxygen), will provide energy to the organism. Under anaerobic conditions, carbon dioxide, hydrogen, and/or methane are typically produced as gases.
The large quantity of CPR materials emplaced in the WIPP is the main carbon source that could lead to substantial gas generation from degradation by microorganisms. As with most subsurface microbial processes, there are large uncertainties surrounding the extent to which microbial consumption of CPR materials can occur during the 10,000-yr WIPP regulatory period. In this context, it is assumed that significant microbial consumption of CPR materials is possible, but this is by no means certain.
To incorporate these uncertainties in the PA, the conceptual model for biodegradation used in CRA-2004 (Wang and Brush 1996a and 1996b) used a probability of 0.50 for significant microbial activity. This was changed in the CRA-2004 PABC calculation to be a probability of 1.0, meaning that microbial activity was considered in all PA vectors. The presence of this microbial activity means it is assumed that microbes may consume 100% of the cellulosic materials in the repository, and that there is a probability of 0.25 that microbes may consume the plastic and rubber materials. Thus, there is microbial consumption of cellulosic materials, but not of plastic or rubber materials, in 75% of the PA realizations (vectors), and microbial consumption of all CPR materials in 25% of the vectors.
Microbial consumption of CPR materials could affect the actinide source term in four ways:
1. Productionof significant quantities of CO2, which could acidify the brine in the absence of an MgO buffer or increase the solubility of actinides by carbonate complexation at the expected mildly alkaline pH
2. Bioreduction of higher-valent actinide species leading to lower-valent, less-soluble actinide species
3. Degradation of solubilizing organic ligands, leading to lower actinide solubility
4. Production of humic and microbial colloids that could increase the amount of actinide pseudocolloids in the brine
The effect of CO2 production is discussed in this section. The remaining three effects are implicitly considered in the analyses that address the oxidation-state distributions (Section SOTERM-4.2), the effects of organic ligands (Section SOTERM-2.3.6), and the effects of colloids (Section SOTERM-3.8). The simplifications used in the PA calculations for all four of these effects are discussed at the end of this section.
Microbial activity, if it occurs to a significant extent in the WIPP, would consume CPR materials by the following sequential reactions (Brush 1990, Francis and Gillow 1994, Brush 1995, Wang and Brush 1996a, and Francis 1998):
C6H10O5 + 4.8H+ + 4.8NO3- ® 7.4H2O + 6CO2 + 2.4N2; (SOTERM.18)
C6H10O5 + 6H+ + 3SO42- ® 5H2O + 6CO2 + 3H2S; (SOTERM.19)
C6H10O5 + H2O ® 3CH4 + 3CO2. (SOTERM.20)
Methanogenesis, described by Reaction (SOTERM.20), is not included as a degradation pathway in CRA-2004 PABC (Leigh et al. 2005) due to uncertainty about the availability of sulfate (see Section SOTERM-2.2.5) in the DRZ and its exclusion is a conservative assumption relative to the amount of carbon dioxide that could be produced. In effect, the CRA-2004 PABC and this PA assume that an excess of sulfate is always available to sustain sulfate-reduction biodegradation pathways. When unlimited sulfate is available from natural sources in the host rock, which is the assumption for the CRA-2004 PABC and for this PA, 4% of the gas generation occurs through denitrification and 96% occurs by way of sulfate reduction (Leigh et al. 2005, Section 2.4).
Microbial consumption of CPR materials, therefore, could produce significant quantities of CO2, which could in turn acidify any brine present in the repository and increase the solubilities of the actinides relative to those predicted for neutral and mildly basic conditions. Therefore, the DOE is emplacing MgO in the repository to decrease actinide solubilities by consuming essentially all of the CO2 that could be produced by microbial consumption of CPR materials, and by buffering (controlling) the fCO2 and pH within ranges that are favorable from the standpoint of actinide speciation and solubility (see Section SOTERM-2.3.2).
Three effects of microbial consumption of CPR materials are recognized in the system performance modeling. A simplification has been made so the effects will be time-independent after 100 years. These effects are
1. CO2 production. With the addition of excess MgO, the effects of CO2 production are minimized, and it is assumed that the system may be modeled using the brucite-hydromagnesite (Mg5(CO3)4(OH)2×4H2O) buffer.
2. Redox effects. After 100 years, the repository will have a reducing environment. This is, in part, established by the postclosure microbial consumption of oxygen, but is also due to the corrosion of steel. This combined effect leads to the formation of an anoxic reducing environment in the WIPP.
3. Production of humic and microbial colloids is possible/probable and likely to be the main colloidal contributor to actinide concentrations in DBR release.
The bioreduction of higher-valent actinides is an important potential effect of microbial activity in the WIPP. This potential effect is beneficial to the WIPP licensing case since it strengthens the current PA assumption that lower-valent, and therefore less-soluble, actinide species will predominate in the WIPP. The bioreduction of actinides has recently been the focus of much research due to its expected role in microbially-mediated remediation and containment of subsurface contaminants (Banaszak, Rittman, and Reed 1998; Banaszak et al. 1999; Lloyd, Young, and Macaskie 2000; Reed et al. 2007; Icopini, Boukhalfa, and Neu 2007; and Francis, Dodge, and Gillow 2008). The extent that this applies to the halophiles typically present in the WIPP is, however, uncertain, although it is expected that similar trends in bioreduction will be observed.
The linkage between actinide oxidation state and microbiological processes for soil bacteria is shown in Figure SOTERM-2. Under anaerobic conditions, U(VI), Pu(V/VI), and Np(V) are reduced for a wide range of microbes and electron donors. U(VI) and Np(V) species are primarily reduced enzymatically by reductases formed. The end product in both of these cases is the actinide in the IV oxidation state. The Pu system, however, is more complex in that there are strongly coupled abiotic and biotic pathways (see Table SOTERM-3) and the formation of Pu(III), rather than Pu(IV), is sometimes observed when Pu(IV) solubilization mechanisms coexist.
Although there is a reasonable expectation that bioreduction of higher-valent actinides will occur for microbiologically active anaerobic systems in the WIPP, WIPP-specific data that support this expectation have not been obtained. For this reason, the potential effects of bioreduction on multivalent actinide systems are not considered in the WIPP PA.
Figure SOTERM-2. Expected Dominant Actinide Oxidation States as a Function of the Standard Reduction Potential at pH = 7 in Water That is in Equilibrium With Atmospheric CO2. The Linkages Between the Redox Potentials, Associated Specific Oxidation States, and Microbial Electron Acceptor Couples are Also Shown (Banaszak, Rittmann, and Reed 1998).
Radiolysis effects in the WIPP are caused by the interaction of ionizing radiation and particles (neutrons, α, β, and γ) with the gases, brines, and materials present in the repository. These effects have not been extensively studied under WIPP-related conditions, but there is a fairly good general understanding of their extent and nature. The strongly reducing and oxidizing transients generated radiolytically in aqueous systems can affect the oxidation state distribution of multivalent metals and actinides. In high–ionic-strength sodium chloride brines, this is primarily exhibited in the oxidation of Am(III) and Pu(III/IV) species to Am(V) and Pu(V/VI). Additionally, the radiolytic breakdown of water leads to the formation of molecular hydrogen, which adds to gas generation in the WIPP after brine inundation. Lastly, radiolytic effects can affect the stability and/or enhance the degradation of waste components (e.g., CPR degradation, iron/metal corrosion, and initial actinide oxidation state distribution) during the unsaturated and saturated phases in repository history.
The effects of radiolysis for most conditions expected in the WIPP are predicted to be transient and insignificant. In this context, there is a recognition that although radiolysis can lead to localized conditions and effects that could oxidize multivalent actinides, the brine chemistry, metal corrosion, and microbiological activity will combine to very rapidly overwhelm these effects. For this reason, radiolysis effects on actinide solubility are not explicitly included in the WIPP PA to calculate actinide concentrations. More specifics on the overall mechanisms, brine radiation chemistry, and potential radiolytic effects on actinide speciation are given in this section.
The radiolysis of high-ionic-strength brine systems has not been extensively studied, but some studies exist (Büppelman, Kim, and Lierse 1988; Kim et al. 1994; Kelm, Pashalidis, and Kim 1999; Ershov et al. 2002). The many components in the brine systems of interest to the WIPP will lead to a relatively complex radiation chemistry and the formation of numerous transients and free radicals.
In contrast to this, the radiation chemistry of pure and dilute aqueous systems has been extensively investigated, and detailed reviews of this research have been published (Draganic and Draganic 1971, Spinks and Woods 1990). The irradiation of pure water leads to the formation of molecular hydrogen peroxide (H2O2) and hydrogen (H2). These molecular yields are relatively insensitive to a wide range of conditions in dilute systems for a given type of ionizing radiation. Molecular yields are GH2 = 0.45 molecule (molec)/100 electron-volt (eV) and GH2O2 = 0.7 molec/100 eV for low Linear Energy Transfer (LET) ionizing radiation (β, and γ) and GH2 = 1.6 molec/100 eV and GH2O2 = 1.5 molec/100 eV for high LET radiation (α and neutrons). The radiolytic formation of hydrogen in the WIPP brine due to self-irradiation effects of 239Pu was established and a molecular yield of GH2 = 1.4 molec/100 eV was measured (Reed et al. 1993). This yield is consistent with the high LET literature, even though the irradiations were performed in brine.
The high concentrations of electron and free radical scavengers present in the WIPP brine have a pronounced effect on the radiation chemistry. Most importantly, halides react with the hydroxyl radical (OH×) or act as scavengers (such as Cl- or Br-) to gradually lower the molecular yield of H2O2 as the concentration of the scavengers is increasing (Kelm, Pashalidis, and Kim 1999). In this context, oxidizing transient species are “chemically” stored as oxychlorides and oxybromides, leading to a shift towards more oxidizing conditions. Figure SOTERM-3 gives an overview of the radiolytic pathways and mechanisms that are likely (Buppelmann, Kim, and Lierse 1988). In NaCl brine, the formation of chloride species (ClO-, HOCl, Cl2, and Cl3-) is favored, instead of H2O2 (Büppelmann, Kim, and Lierse 1988).
Kelm, Pashalidis, and Kim (1999) showed that the formation of hypochlorite ion increases with the chloride concentration and the dose (Figure SOTERM-4) in NaCl brine. The authors found that in solutions containing 37 gigabecquerel (GBq)/liter (L) of 238Pu, the hypochlorite concentration increases with time (dose) and appears to approach a steady state (see Figure SOTERM-4). At a constant dose rate, the maximum hypochlorite concentration depends on the chloride concentration. It was also observed that hypochlorite ion generation was negligible when chloride concentrations were smaller than 2 M.
In the WIPP brine, however, some solutes other
than chloride may play a role. Ershov et al. (2002) showed that small amounts of bromide in natural brines
under radiolysis can give Cl2-,
ClBr-, and Br- radical anions at the
radical step, and then mixed halogen molecules and trihalide ions
by radical recombination at the molecular step (Ershov et al.
2002). The hydrolysis of
Figure SOTERM-3. NaCl Brine Radiolysis Species and Suggested Mechanism of Production. The Formation of Chloride Species (ClO-, HOCl, Cl2, and Cl3-) is Favored Instead of H2O2 (Based on Data in Büppelmann, Kim, and Lierse 1988).
Figure SOTERM-4. Radiolytic Formation of Hypochlorite Ion in Solutions of Various NaCl Concentrations at a Constant Alpha Activity of 37 GBq/L at pH~12 (Based on Data in Kelm, Pashalidis, and Kim 1999)
mixed halogen molecules can then result in the formation of hypobromite (OBr-) (acidic form: hypobromous acid [HOBr]), a starting substance to more stable bromates of higher oxidation state (Ershov et al. 2002).
Some WIPP-specific experiments were performed to establish the key radiolytic product in GWB and ERDA-6 brine (Lucchini et al. 2009). This study confirms that hydrogen peroxide (H2O2) and hypochlorite ion (OCl-) are unstable in these WIPP brines, due in part to metallic impurities in the brine. There was, however, an accelerated decomposition of these species when bromide (Br-) was present, which is the case for both ERDA-6 and GWB brines. Here, OCl- readily and stoichiometricly reacted with Br- to form hypobromite ion (OBr-), which appeared to be the most important radiolytic transient observed under these conditions. OBr-, like OCl-, is also an oxidizing species (Eº=0.76V), that will likely lead to the oxidation of multivalent actinides in the WIPP, but this reactivity has not been established experimentally under representative WIPP conditions (Lucchini et al. 2009).
In the WIPP, most of the brine radiolysis is caused by the deposition of alpha particles from the TRU isotopes present in the WIPP waste. The range (distance traveled until the alpha particle’s energy is lost) of these alpha particles is very short (<40 microns) and radiolysis of the brine solution will take place at the solid-liquid interface. Locally, the concentration of oxidative radiolytic products of brine, such as hypochlorite, chlorite, chlorate, and products of their reaction with brine components (e.g., hypobromite) may be high, and they may directly interact with the radioactive surface. These “very-near” radiolytic effects, however, are expected to be quickly mitigated by the bulk brine chemistry and the reaction of reducing agents (e.g., reduced iron) with the oxidizing molecular products formed.
A buildup of oxidizing radiolytic products in brine may increase the redox potential of the brine (Büppelmann, Kim, and Lierse 1988), and consequently directly generate higher-valent actinide species. Alternatively, these radiolytic products could be inserted into some solid actinide phases. For example, Kim et al. (1994) studied the solubility of schoepite, (UO2)(OH)2×xH2O, with hypochlorite ion in 0.1M NaCl at 25 °C (77 °F), in CO2-free atmosphere (Kim et al. 1994). Their X-Ray Diffraction (XRD) patterns of the residual precipitates showed the introduction of hypochlorite ion in precipitates. Kim et al. (1994) observed that the presence of hypochlorite ion in the initial schoepite structure enhanced the solubility of the solid 10 to 100 times in the range of pH 6.0-9.8, compared with its solubility in the absence of hypochlorite ion (Kim et al. 1994). However, this effect was reduced when the molar ratio [ClO-]/[UO22+] increased. This scenario is unlikely to occur in the WIPP because the potential buildup of oxidizing radiolytic products generated in brine is readily overwhelmed by the overall reducing capacity of the site (reduced metals and microbial processes).
The buildup of oxidizing radiolytic products due to brine radiolysis has also been shown to significantly affect the solution chemistry of Am. For example, Am(III) was oxidized to the more soluble forms of Am, namely AmO2+ and AmO22+ (Magirius, Carnall, and Kim 1985; Katz, Seaborg, and Morss 1986; Stadler and Kim 1988; and Meyer et al. 2002). Magirius, Carnall, and Kim (1985) reported on the radiation effects exerted upon a 5 M NaCl solution at the pH 8 to 9 range using precipitated Am(OH)3 at a concentration of 1.03 × 10-3 M (1.07 curie [Ci]/L). They observed that the precipitate began to show discoloration, changing from pink Am3+ to brown AmO2+, within 24 hours (h), with quantitative oxidation of all the Am to AmO2+ within 1 week. Because Pu is more readily oxidized than Am, the expectation is that Pu could also be oxidized in irradiated brine. The metastability of Pu(VI) in the WIPP brine when no reducing agents were present was established and attributed to self-radiolysis effects of the 239Pu isotope used (Reed, Okajima, and Richmann 1994; Reed et al. 2006).
Stadler and Kim (1988) also report the existence of higher oxidation states of Am, due to self radiolysis. Solubility experiments on Am(OH)3(solid[s]) in 3 M NaCl resulted in much higher Am concentrations than was calculated from the solubility product. This difference was assigned to the radiolytic oxidation of Am3+ to AmO2+. Spectrophotometric evidence of AmO2+ species in solution was reported. The authors report the value of log10KS,0 = -9.3 ± 0.5 for the reaction
AmO2OH(s) D AmO2+ + OH- (SOTERM.21)
The solubility product of AmO2OH(s) is in general agreement with other solubility studies on different pentavalent actinides.
These results show there is clearly a potential for oxidized, higher-valent actinides to form in brine when no reducing agents are present. This, however, needs to be interpreted in the context of the strong reducing agents and processes that will predominate in the WIPP, such as bioreduction (Section SOTERM-2.4.1.2), iron reduction (Section SOTERM-3.4.2), and reduction by organic complexants (Section SOTERM-2.3.4). WIPP-specific data show that the presence of reduced iron (Fe(II/0)) leads to a rapid reduction of Pu(VI) to Pu(IV) species under a wide range of anoxic conditions (Reed et al. 2006, Reed et al. 2009). These results are expected to extend to the Am(V) system, since this species is more readily reduced than Pu(V/VI). Reduced iron will also react with radiolytically generated oxidizing species, such as hypochlorite or hypobromite, to prevent their buildup in the brine solution with time. In summary, these WIPP-specific results show that the reductants present in WIPP waste (reduced metals and organics) will overwhelm potential radiolytic effects under the expected conditions in the WIPP, and a significant and sustained radiolytic enhancement of actinide solubilities is not predicted.
There are no significant changes in the WIPP repository conditions, chemistry, and processes since the CRA-2004 and the last PA performed (Leigh et al. 2005). Specifically, the assumptions and parameters given in Table SOTERM-1 are the same as those used for the CRA-2004 PABC. This applies to all the discussions in Section SOTERM-2.1, Section SOTERM-2.2, Section SOTERM-2.3, and SOTERM-2.4.
Three WIPP-relevant processes were reviewed and updated. First, actinide reduction is a direct consequence of microbial activity under anoxic conditions (see Section SOTERM-2.4.1). This, in fact, strengthens the current PA position that reducing conditions will be maintained in the WIPP, although this is not accounted for in PA. Second, there is an increased understanding of the effects of ionizing radiation on actinide speciation in sodium chloride brine systems (Section SOTERM-2.4.2). This leads to recognition of the role of hypobromite in WIPP-specific brines. Additionally, WIPP-specific data show that the effects of radiolysis, which can create locally more oxidizing zones in the repository, are readily overwhelmed by the effects of reduced iron. Third, further progress was made in understanding the reaction sequence and interactions of MgO in WIPP-relevant brine systems (see Section SOTERM-2.3.3).
The TRU inventory was updated. This update impacted the concentration of organic chelating agents (see Section SOTERM-2.3.6), the volume of the TRU waste inventory, and the amount of emplaced materials which increased the CPR inventory (see Section SOTERM-2.3.7).
It is important to note that the CRA-2004 PABC included changes to the CRA-2004 PA in response to comments received from the EPA (Cotsworth 2005). These are discussed in detail as part of the CRA-2004 PABC documentation (Leigh et al. 2005). The specific changes for CRA-2004 PABC that revised or clarified the microbial assumptions and input are as follows:
1. Gas generation rates were revised to account for slower, longer-term processes based on new WIPP-specific data (Nemer and Stein 2005; Nemer, Stein, and Zelinski 2005).
2. All PA vectors are now assumed to be microbial (this was changed from a probability of 0.5 in CRA-2004 PA).
3. Gas generation is assumed to occur only through denitrification (~4%) and sulfate reduction (~96%). It is assumed that methanogenesis does not occur because sulfate is assumed to be always available for microbial processes.
The speciation of actinides under WIPP-relevant conditions defines the source term for actinide release from the WIPP in release scenarios where dissolved actinide concentrations are important (e.g., DBR and transport through the Salado or Culebra). The key factors that establish the concentrations of dissolved actinides under subsurface conditions are known. The most important of these factors for the WIPP repository are listed below.
1. Actinide redox chemistry is a critical factor in establishing the concentration of actinides in brine. The solubility of reduced actinides (III and IV oxidation states) is significantly lower than oxidized forms (V and/or VI). In this context, maintaining reducing conditions in the WIPP and the strong coupling of the chemistry for reduced metals and microbiological processes with actinides are important.
2. The complexation of each actinide species is a critical factor in defining its solubility. For a given oxidation state, the inorganic and organic complexes present will define the solubility of the actinide. These complexants are in the preemplacement environment, are part of the TRU waste that is emplaced, or are produced as a result of subsurface processes, most notably microbial and corrosion processes.
3. Intrinsic and pseudoactinide colloid formation is a critical factor in defining the overall solution concentration of each actinide. The contribution of actinide colloids to the concentration of actinides in the WIPP is predicted to be significant. Many of the key TRU species in their expected oxidation states tend to form colloids or strongly associate with nonactinide colloids present (e.g., microbial, humic and organic).
The WIPP PA approach as established in the initial WIPP license application (U.S. Department of Energy 1996) and continued through the most recent PA calculations (Leigh et al. 2005) accounts for all three of these key factors.
The PA concept of actinide speciation in the WIPP is well grounded in what has been observed for actinide contaminants in near-surface groundwater. In natural systems, the following inorganic ligands are potentially important complexants of radionuclides in solution: CO32-/HCO3-, OH-, C1-, SO42-/S2-, fluoride (F-), and phosphate. Additionally, anthropogenic and bioderived chelating agents can strongly bind actinide species and will compete with the inorganic complexants present. Lastly, the tendencies of actinides to form intrinsic colloids and strongly associate or bind with colloidal particles are also well established. The relative importance of these complexants and processes depends on the pH, radionuclide oxidation state present, the presence of other metals, and the relative ligand concentrations. There are a number of general reviews on various aspects of actinide environmental chemistry (Allard 1982; Choppin, Liljenzin, and Rydberg 2004 [pp. 94–112]; Clark, Hobart, and Neu 1995; Banaszak, Rittmann, and Reed 1998; Runde 2000; Nitsche et al. 1992).
For the anoxic, reducing, and mildly basic brine systems expected in the WIPP (see Table SOTERM-1), the most important inorganic complexants are expected to be carbonate/bicarbonate and hydroxide. There are also important organic complexants that coexist in TRU waste with the potential to strongly influence actinide solubility. In this context, the relative importance of actinides and overall oxidation state, based on the CRA-2004 PABC TRU waste inventory, with respect to the potential release of actinides from the WIPP, is:
Actinides: Pu ≈ Am >> U > Th >> Np ≈ Cm (SOTERM.22)
Actinide Oxidation State: An(III) > An(IV) >> An(VI) >> An(V) (SOTERM.23)
In the CRA-2004 PABC (Leigh et al. 2005), the contribution of Pu, Am, U, Th, Cm, and Np is expressly considered, although only Pu and Am contribute significantly to TRU release from the WIPP. The III oxidation state is the most important oxidation state based on current WIPP PA assumptions because Am always exists in the III state, Pu exists in the III state in 50% of the vectors, and the III oxidation state is more soluble than the IV (see Section SOTERM-4.0 for a more detailed discussion).
In this section, an update of the literature and a summary of new WIPP-specific data is provided (when available) for all the actinides that contribute in one way or another to the PA. Section SOTERM-3.1 gives an overview of the projected and current inventory of actinides in the WIPP; Section SOTERM-3.2, Section SOTERM-3.3, Section SOTERM-3.4, Section SOTERM-3.5, and SOTERM-3.6 contain an overview of the relevant environmental chemistry and WIPP-specific results for Th, U, Np, Pu, and Am/Cm, respectively; Section SOTERM-3.7 pertains to the complexation of actinides by organic chelating agents in the WIPP; Section SOTERM-3.8 provides an overview of the potential for the formation of actinide colloids in the WIPP; and Section SOTERM-3.9 is a summary of changes since the CRA-2004 and CRA-2004 PABC. An up-front overview of these sections appears in Table SOTERM-6. The PA implementation of this actinide environmental chemistry is discussed in Section SOTERM-4.0 and Section SOTERM-5.0.
The actinide inventory for the WIPP, based on the Transuranic Waste Baseline Inventory Report-2004 inventory (U.S. Department of Energy 2006), is given in Table SOTERM-7. This is also the inventory used to calculate the CRA-2004 PABC (Brush and Xiong 2005b) actinide solubilities. Also included in this table are the calculated inventory limits of the various actinides and radionuclides considered by the WIPP PA.
Over long time frames, only Pu and Am are expected to make a significant contribution to releases from the WIPP. Curium (Cm), which is predominantly present as 244Cm, is a factor of 10 below the calculated solubility for III actinides when fully dissolved and, with its very short half-life (18.11 years), will not be important beyond the 100-year period of institutional control. Although relatively large inventories of cesium (Cs) and strontium (Sr) are projected, these can only contribute significantly to the overall release from the WIPP for the first 100 years of repository history, so are not significant beyond the period of institutional control.
Table SOTERM-8 gives the panel-specific inventory of the
actinides for Panels 1 and 2 in the WIPP. These data are
based on characterization of containers in Panels 1 and 2 WIPP Waste Information System
(WWIS). Also included in this inventory is the amount of
key waste
Table SOTERM-6. Overview of the WIPP PA View/Role and Relevant Environmental Chemistry of the Key Actinide Species in the WIPP (References for Each Actinide are Provided in the Following Sections)
|
Actinide |
WIPP PA View/Role |
Environmental Chemistry |
|
Thorium |
Not a TRU component. Currently included in PA calculations, but not a significant contributor to actinide release. Used as an oxidation-state invariant analog for the IV actinides. Th data are used in Fracture-Matrix Transport (FMT) to calculate the solubility of Pu(IV), Np(IV), and U(IV). |
Exists as Th4+ complexes and is sparingly soluble under a wide range of environmental conditions. |
|
Uranium |
Not a TRU component. Potentially useful as a VI analog for Pu(VI) species. Currently, U is conservatively assumed to be U(VI) in 50% of the PA vectors (set at a 1 mM solubility) and U(IV) in 50% of the PA vectors. |
Exists as UO22+ and U4+ species that are strongly correlated with redox conditions. Can form highly insoluble U(VI) and U(IV) phases. Can persist up to mM concentrations in near-surface groundwater. |
|
Neptunium |
TRU component. Currently included in PABC calculations, but not a significant contributor to actinide release. Assumed to be IV in 50% of the PA vectors and V in 50% of the PA vectors. Expected to be in the IV oxidation state under the conditions expected in the WIPP. |
Mobile and relatively soluble as the NpO2+ species under oxidizing conditions. Is fairly insoluble and immobile as Np4+ under reducing conditions. |
|
Plutonium |
TRU component. Major contributor to actinide release calculations. Assumed to be IV in 50% of PA vectors and III in the other 50% of PA vectors. |
Relatively immobile and insoluble as a subsurface contaminant. Persists as Pu4+ except under biomediated, strongly reducing conditions where transitory Pu3+ species may be formed. Expected to be transported primarily through colloidal mechanisms. |
|
Americium |
TRU component. Major contributor to actinide release calculations. Exists in the III oxidation state in all vectors and its thermodynamic data is used by FMT for all III oxidation state calculations. Significant colloidal contribution due to strong association as a pseudocolloid. |
Relatively immobile and insoluble as a subsurface contaminant. Persists as Am3+ complexes under a wide range of environmental conditions. |
|
Curium |
Small quantities of 243Cm, 245Cm, and 248Cm are present in the WIPP. 244Cm, although present, is not a TRU waste component due to its <20 year half-life. These are very minor contributors to actinide release. Chemistry is analogous to Am(III). |
Not a very significant concern as a subsurface contaminant. Has the same chemistry as Am, so it will persist as a Cm3+ species. |
|
Organic Chelating Agents |
The effects of EDTA, citrate, oxalate, and acetate on actinide solubility are considered in WIPP PA. These are present in WIPP waste and it is assumed that they are neither destroyed nor created by WIPP-relevant subsurface processes. |
EDTA can persist under a wide range of environmental conditions and strongly chelates actinides. Citrate, oxalate, and acetate will likely be degraded due to microbial activity. |
|
Actinide Colloids |
Pseudocolloids with actinides are formed. These are accounted for in WIPP PA and add to the conservatism of the actinide concentrations calculated. |
Importance and role of An colloid-facilitated transport are the subject of much ongoing debate. Although colloids are formed, it is not clear that they lead to increased actinide migration. |
Table SOTERM-7. WIPP Radionuclide Inventory (U.S. Department of Energy 2006) Decay-Corrected to 2002. This Inventory was the Basis of CRA-2004 PABC Calculations.
|
Radionuclide |
Activity (Ci) |
Amount (kg) |
Element-Specific Inventory |
Inventory-Defined Potential Solubilitya (M) |
|
Actinides |
||||
|
229Th |
1.55E+00 |
7.82E-03 |
5.07 Ci 3.11E+04 kg |
>> Solubility |
|
230Th |
9.72E-02 |
4.71E-03 |
||
|
232Th |
3.42E+00 |
3.11E+04 |
||
|
233U |
1.23E+03 |
1.27E+02 |
1.68E+03 Ci 6.47E+05 kg |
>> Solubility |
|
234U |
2.27E+02 |
3.65E+01 |
||
|
235U |
4.99E+00 |
2.31E+03 |
||
|
236U |
2.78E+00 |
4.29E+01 |
||
|
238U |
2.17E+02 |
6.45E+05 |
||
|
237Np |
6.89E+00 |
9.77E+00 |
6.89 Ci 9.77 kg |
4 ´ 10-6 M (≥ projected solubility) |
|
238Pu |
1.45E+06 |
8.49E+01 |
4.22E+06 Ci 9.93E+03 kg |
>> Solubility |
|
239Pu |
5.83E+05 |
9.40E+03 |
||
|
240Pu |
9.57E+04 |
4.20E+02 |
||
|
241Pu |
2.09E+06 |
2.03E+01 |
||
|
242Pu |
1.27E+01 |
3.23E+00 |
||
|
244Pu |
5.53E-03 |
3.10E-01 |
||
|
241Am |
4.89E+05 |
1.42E+02 |
4.89E+05 Ci 143 kg |
6 ´ 10-5 M (≥ projected solubility) |
|
243Am |
7.88E+01 |
3.95E-01 |
||
|
244Cm |
7.26E+03 |
8.97E-02 |
7.26E+03 Ci 0.0897 kg |
4 ´ 10-8 M |
|
Fission Productsb |
||||
|
137Cs |
4.33E+05 |
5.02E+00 |
4.33E+05 Ci 5.02 kg |
4 ´ 10-6 M |
|
90Sr |
3.78E+05 |
2.77E+00 |
3.78E+05 Ci 2.77 kg |
3 ´ 10-6 M |
|
a Moles in the inventory divided by the minimum brine volume (10011 m3) b Fission products are not TRU, but are considered in the PA to calculate overall release |
||||
Table SOTERM-8. Total Amount (in Kilograms) of Key Waste Package Components and Actinides Present in WIPP Panels 1 and 2 (Based on Data in Lucchini et al. 2007)
|
Panel 1 |
||||
|
Radionuclides |
Amount in kg (Ci) |
Materials |
Amount (kg) |
|
|
241Am |
34.6 (1.19 ´ 105) |
Iron-based metal alloys |
3,327,871 |
|
|
Pu (total) |
2,571 |
Aluminum-based metal alloys |
5,459 |
|
|
239Pu |
2,416 (1.5 ´ 105) |
Other metal alloys |
46,793 |
|
|
U (total) |
22,232 |
MgO |
4,482,355 |
|
|
238U |
22,170 (7.5) |
Cellulosics |
706,141 |
|
|
237Np |
0.6 (0.42) |
Plastic |
522,688 |
|
|
Panel 2 |
||||
|
Radionuclides |
Amount in kg (Ci) |
Materials |
Amount (kg) |
|
|
241Am |
9.2 (3.2 ´ 104) |
Iron-based metal alloys |
4,922,035 |
|
|
Pu (total) |
1,405 |
Aluminum-based metal alloys |
17,730 |
|
|
239Pu |
1,306 (8.1 ´ 104) |
Other metal alloys |
121,526 |
|
|
U (total) |
6,850 |
MgO |
6,667,625 |
|
|
238U |
6,808 (2.3) |
Cellulosics |
477,213 |
|
|
237Np |
1.2 (0.85) |
Plastic |
876,399 |
|
components emplaced. From the perspective of actinide solubility and PA, the most important of these are MgO and iron. Over 8,000 metric tons of iron is already emplaced, contrasting with the much smaller amounts of TRU present (1.8 kg of Np, 43.6 kg of Am, and 3.8 metric tons of Pu). Approximately 11,000 metric tons of MgO are present. These data support and are consistent with current WIPP PA assumptions that sufficient MgO and an overwhelming amount of iron will be present in the WIPP to establish strongly reducing conditions and favorable carbonate levels.
Th is not a TRU component. An estimated 31 metric tons of Th will be in the WIPP. The release of Th as the 230Th isotope was calculated in the CRA-2004 PABC and does not significantly contribute to the overall release of activity from the WIPP. Th is, however, important for the WIPP in that it is used as a redox-invariant analog for the IV actinides (Pu(IV), Np(IV), and U(IV)), and Th complexation data is used in the FMT code for the An(IV) solubility calculations (see Section SOTERM-4.4.3).
Th, under a wide range of conditions, has one stable oxidation state in aqueous solutions: the Th4+ tetravalent ion. For this reason, the environmental chemistry of Th is understood from the perspective of the solubility and complexation of this species, which is also the species expected to be present in the WIPP environment when DBR and transport release scenarios are important.
Other oxidation states for Th in aqueous systems have been reported. Recent data by Klapötke and Schulz (1997) that suggests a Th3+ species as a somewhat stable species in slightly acidic solution is not correct; it has been discounted because the proposed reaction for the species’ formation is shown to be thermodynamically impossible, and the azido-chloro Th4+ complex is incorrectly assigned to the Th3+ species (Ionova, Madic, and Guillaumont 1998).
The hydrolysis of Th4+, as is true for all An(IV) species in the WIPP, is complex and a critically important interaction in defining the overall solubility of Th. This was recently investigated by Ekberg et al. (2000), Rai et al. (2000), Moulin et al. (2001), and Okamoto, Mochizuki, and Tsushim (2003) and critically reviewed by Neck and Kim (2001) and Moriyama et al. (2005). The authors have proposed a comprehensive set of thermodynamic constants that extends to all tetravalent actinides. The solubility products were determined for amorphous (am) Th(OH)4 (Neck et al. 2002, Altmaier et al. 2005; Altmaier et al. 2006) and for crystalline ThO2 (Neck et al. 2003), as well as for specific ion interaction theory parameters (Neck, Altmaier, and Fanghänel 2006). The thermodynamic stability constants are listed in Table SOTERM-9.
Table SOTERM-9. Thermodynamic Stability Constants for Key Th Hydrolytic Species
|
Hydrolytic Reaction/Species |
Stability Constant |
|
Mononuclear Species |
|
|
Th(OH)4, am D Th4+ + 4OH- Th(OH)4, cr D Th4+ + 4OH- Th4+ + OH- D Th(OH)3+ Th4+ + 2OH- D Th(OH)22+ Th4+ + 3OH- D Th(OH)3+ Th4+ + 4OH- D Th(OH)4,aq |
log Ks,am = -47.8 ± 0.3 log Ks,cr = -53.2 ± 0.4 log β01 = 11.8 ± 0.2 log β02 = 22.0 ± 0.6 log β03 = 31 ± 1 log β04 = 38.5 ± 1 |
|
Polynuclear Species |
|
|
4Th4+ + 12OH- D Th4(OH)124+ 6Th4+ + 15OH- D Th6(OH)159+ |
log β04,12 = 141 log β06,15 = 176 |
The presence of carbonate in solution greatly increases the solubility of thorium dioxide (ThO2). An increase by one order of magnitude of the carbonate concentration in the range of 0.1 – 2 M leads to a five-order-of-magnitude increase in the Th(IV) solubility due to the formation of mono- and penta-carbonate complexes. Östhols, Bruno, and Grenthe (1994) proposed the following equilibrium reactions and the corresponding stability constants:
ThO2 + H+ + H2O + CO32- D Th(OH)3 CO3- log K131 = 6.11 ± 0.19 (SOTERM.24)
ThO2 + 4H+ + 5 CO32- D Th(CO3)56- + 2H2O log K105 = 42.12 ± 0.32 (SOTERM.25)
This speciation scheme, however, has been criticized in recent work (Altmaier et al. 2005) because it overpredicts the dependency of Th solubility on carbonate and underpredicts the effect of hydrolysis at higher pH. That hydrolysis prevails at pH >10 is supported by relatively detailed experimental results (see Figure SOTERM-5). These data are explained by the predominance in this system of Th(OH)(CO3)45- complex rather than Th(CO3)56-. A greater role for other ternary complexes of thorium (e.g. Th(OH)2(CO3)22-), which are also likely to be present in the WIPP conditions, is also proposed, and formation constants for these complexation reactions are reported. The use of the pentacarbonyl complex for the IV actinides in the WIPP PA, for these reasons, is a conservative assumption that overpredicts the solubility of the IV oxidation state at pH > 10. A correction in the FMT database to the value of the Th(OH)4(aqueous [aq]) to be consistent with Neck et al. (2002) was incorporated into the CRA-2004 PABC.
Oxyanions such as phosphate and, to a lesser extent, sulfate, also form Th4+ complexes that can precipitate at pH <5. The effect of phosphate on solubility of microcrystalline ThO2 is very limited. The stability constants for Th4+/H2PO4- and Th4+/HPO42- were reported (Langmuir and Herman 1980). Overall, the role of these oxyanions is expected to be unimportant for the mildly basic brines (pH ~8-10) present in the WIPP.
A new perturbation to the understanding of Th speciation, as well as other actinides in the IV oxidation state, is the recent observation that Ca, and to a lesser extent, magnesium (Mg), enhances Th solubility at pH >10 when carbonate is present. In recent publications, the formation of Ca4[Th(OH)8]4+ and Ca4[Pu(OH)8]4+ ion pairs in alkaline CaCl2 solution is reported (Brendebach et al. 2007; Altmaier, Neck, and Fanghänel 2007). These species cause a rapid increase in the solubility of all tetravalent actinides at pH greater than 11. This increased solubility is only observed at CaCl2 concentrations above 0.5 M for Th(IV), and correspondingly above 2 M for Pu(IV) species. This effect can be discounted for the WIPP PA because Ca concentrations in the WIPP are predicted to be approximately 14 mM or less with a pH of approximately 8.7. These are both well below the levels needed to see a significant effect for both Th and Pu.
Actinides in the IV oxidation state, because of the complexity of their solution chemistry and very high tendency towards hydrolysis, form colloidal species in groundwater. The potential effect of colloid formation on solubility of Th(IV) in concentrated NaCl and MgCl2 solution was recently published by Altmaier, Neck, and Fanghänel (2004) and is shown in Figure SOTERM-6. In neutral-to-alkaline solutions, colloids could be formed as Th oxyhydroxide with log [Th](colloid [coll]) = -6.3 ± 0.5, independent of ionic strength. In Mg solutions, the formation of pseudocolloids (i.e., Th(IV)) sorbed onto Mg2(OH)3Cl·4H2O(coll) led to an apparent increase of the total Th concentration up to 10-5 M (Walther 2003, Degueldre and Kline 2007, Bundschuh et al. 2000). For these reasons, colloid formation is addressed in the WIPP PA.
There were no new WIPP-specific data on Th
solubility and speciation obtained since CRA-2004. There
were, however, a number of WIPP-relevant experiments reported in
simplified brine systems, mainly by researchers at
Karlsruhe. These results were summarized above in the
context of their relationship to the environmental chemistry of
Th in high-ionic-strength systems. These solubility data
support the current WIPP PA assumptions on An(IV) solubility and
extend
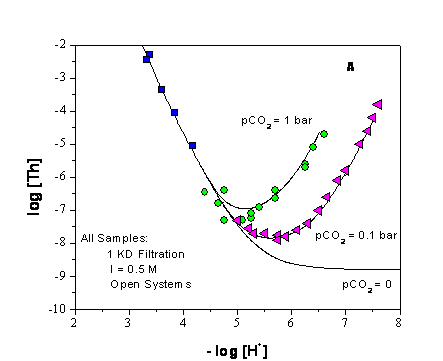
Figure SOTERM-5. Solubility of Amorphous Th(IV) Oxyhydroxide as a Function of Carbonate Concentration in 5 M for pH + 2–8 (A) and pH = 8–13.5 (B). The Solid Lines are the Calculated Solubilities (Based on Data in Altmaier et al. 2005).
Figure SOTERM-6. Solubility of Th(OH)4(am)Determined from Undersaturation in 0.5 NaCl, 5.0 M NaCl, and 2.5 M MgCl2. Filled Points: Total Th Concentrations (Including Colloids); Open Points: Th Concentrations Measured after Ultracentrifugation at 90,000 Revolutions Per Minute (5 × 105 g) (Based on Data in Altmaier, Neck, and Fanghänel 2004).
past project data to a broader range of pH and carbonate levels. These results also note that Ca-enhanced carbonate complexation, something that has only been understood in the last couple of years, can greatly increase the solubility of IV actinides. This complexation, however, requires relatively high pH in combination with very high Ca levels, something that is not expected in the WIPP. The expected pH and dissolved Ca levels predict little or no effect due to this complex.
U is not a TRU component, but is, by mass, expected to be the most prevalent actinide component in the WIPP. Current estimates predict that ~647 metric tons will be placed in the repository (see Table SOTERM-8). By mass, greater than 99% of this U will be the 238U isotope, with minor amounts of 233U, 234U, 235U, and 236U. U does not contribute significantly to the calculation of actiniderelease through cuttings/cavings and spallings because of its low specific activity. U release can occur through the Culebra in very small amounts because of its potentially high solubility in the VI oxidation state.
U release, as the 234U isotope, was calculated in the CRA-2004 PABC. In the WIPP PA, the oxidation state distribution assumption is that U speciates as U(IV) in 50% of the PA vectors and as U(VI) in the other 50% of the vectors. The U concentration for this oxidation state is currently set at 1 mM (U.S. Environmental Protection Agency 2005), since there is no An(VI) model in the WIPP. U(IV) solubility is calculated using the Th(IV) speciation data reported in the FMT model. For the current WIPP PA assumptions, uranium does not contribute significantly to the overall release of actinides from the WIPP.
U is by far the most studied of the actinides under environmentally relevant conditions. An extensive review of this chemistry, as it relates to the WIPP case, was completed (Lucchini et al 2009) and more general reviews can be found (Morss, Edelstein, and Fuger 2006, Guillaumont et al. 2003). An overview of U environmental chemistry is presented in this section.
U can theoretically exist in aqueous solution in the III, IV, V, and VI oxidation states (Hobart 1990; Keller 1971 [pp. 195–215]; Clark, Hobart, and Neu 1995). In the environment, however, only the IV and VI oxidation states, which exist as U4+ and UO22+ species, are present. U3+, should it be formed, is metastable and readily oxidized in aqueous solution, and U(V) only exists as a very short-lived transient that instantaneously disproportionates to form U(IV) and U(VI) species. The corresponding reduction potential diagram for U at pH = 0, 8, and 14 is given in Figure SOTERM-7 (Morss, Edelstein, and Fuger 2006).
Figure SOTERM-7. Reduction Potential Diagram for U at pH = 0, 8, and 14 (Based on Data in Morss, Edelstein, and Fuger 2006). For the Expected Reducing and Mildly Basic pH Conditions in the WIPP, U(IV) is Predicted to be the Predominant Oxidation State.
Under oxidizing subsurface conditions typical of most near-surface groundwaters, U(VI) as UO22+ uranyl complexes, is the predominant oxidation state and is not easily reduced geochemically. Thermodynamically, uranyl species are stable even under mildly reducing conditions and are not reduced by some Fe(II) phases (see Table SOTERM-3). In anoxic WIPP brine experiments with a hydrogen overpressure, uranyl persists as a stable hydrolytic or carbonate complex for over two years (Reed and Wygmans 1997).
In the anoxic and strongly reducing environment expected in the WIPP, however, potential reduction pathways exist. The two most important of these reduction pathways are reaction of uranyl with reduced iron phases (Fe[0/II]), and bioreduction through enzymatic pathways by anaerobic microbes, such as metal reducers, sulfate reducers, and methanogens (see Figure SOTERM-2). For these reasons, U(IV) is the oxidation state expected to predominate in the WIPP when brine inundation occurs.
The use of iron barriers in the removal of uranyl from groundwater is well established and has been reported for the removal of U(VI) from groundwater using zero-valent iron barriers (Gu et al. 1998, Fiedor et al. 1998, Farrell et al. 1999) and iron corrosion products formed in saline solution (Grambow et al. 1996). However, in those studies, it was unclear whether the removal of uranyl (UO22+) resulted from reductive precipitation or from adsorption onto the iron corrosion products (Gu et al. 1998). In their experiments under saline conditions, Grambow et al. (1996) found that a large percentage of U was rapidly adsorbed onto the iron corrosion products consisting of over 97% hydrous Fe(II) oxide, and very little U(IV) was found. The complexity of the U-Fe-H2O-CO2 system explains the scarcity of the experimental data and the lack of a predominant mechanism (reduction-precipitation or adsorption) for the removal of U(VI) in the presence of iron.
Under anoxic conditions, Trolard et al. (1997) establishes that the corrosion of steel and iron generates Fe(II)/Fe(III) hydroxide species known as green rusts. Green rusts contain a certain amount of nonhydroxyl anions (carbonate, halides, or sulfate); they have a high specific surface area (Cui and Spahiu 2002) and a high cation sequestration capacity (O’Loughlin et al. 2003). They are considered metastable oxidation products of Fe(II) to magnetite Fe3O4 and Fe(III) oxyhydroxides (e.g., goethite α-FeOOH) (O’Loughlin et al. 2003). A few experimental studies demonstrate that U(VI) is reduced to U(IV) by green rusts (Dodge et al. 2002, O’Loughlin et al. 2003). The formation of a UO2 phase was measured by Extended X-Ray Absorption Fine Structure (EXAFS) analysis (O’Loughlin et al. 2003) or by X-Ray Absorption Near Edge Structure (XANES) analysis and confirmed by X-ray Photoelectron Spectroscopy and Fourier Transform Infrared Spectroscopy (FTIR) (Dodge et al. 2002).
Banaszak, Rittman, and Reed (1998) have reviewed the important role of microbial processes in the reduction of multivalent metals under anaerobic/reducing conditions. For uranyl in particular, several studies exist that show that U(VI) is reduced to U(IV) species under a wide range of conditions (Lovley et al. 1991, Lovley et al. 1993, Barton et al. 1996, Huang et al. 1998, Abdelouas et al. 2000, Bender et al. 2000, Fredrickson et al. 2000). Most of this work pertains to groundwater bacteria, and is not directly applicable to the WIPP.
There are relatively few studies that investigate the interaction of U with the halophiles that are more typically present in WIPP brine. Some WIPP-relevant research was done (Francis et al. 2000), but this work was mostly focused on gas generation, not actinide interactions. It remains to be demonstrated that the mechanisms leading to the bioreduction of U(VI) also extend to the microbes present in the WIPP, although it is fully expected that this will be the case.
Tetravalent U is expected to be the dominant oxidation state in the WIPP as a result of the reducing conditions that will prevail. The solubility of U(IV) under these conditions is analogous to that observed for Th (see Section SOTERM-3.2 and discussion in Section SOTERM-4.1) and is, in fact, calculated in the WIPP PA with the Th(IV) database.
Under strictly reducing conditions, such as those expected in the WIPP, the most common and stable U solid is U dioxide (UO2, uraninite) and associated hydrates. The aqueous species that predominates between pH 5-10, at low carbonate concentration, is the neutral tetra-hydroxide complex. This is described by the following equation:
U4+ + 4OH- ↔
U(OH)4(aq) ↔ UO2(s) +
2H2O ↔ UO2×xH2O×(s) +
(2-x)H2O
(SOTERM.26)
The solubilities reported in the literature for hydrous UO2×xH2O(s), amorphous (am) UO2, or microcrystalline UO2 are very scattered (Neck and Kim 2001). The explanation in the NEA review on U speciation (Guillaumont et al. 2003) is that the solubility data reported do not correspond to a unique material, but rather to a range of U oxide solids that have different thermodynamic stabilities. The species predominance diagrams for U are shown in Figure SOTERM-8 for aqueous and solid phases (Casas et al. 1998), based on the thermodynamic constants given for U at 25 ºC by Guillaumont et al. (2003).
Experimentally, U4+ is readily oxidized to UO22+. This occurs even when only trace levels of oxygen, which are often below the limit of detection by most laboratory instrumentation, exist. This explains why there are relatively few studies of U4+. It is also problematic because there are very large discrepancies in the literature as a result of experimental artifact. In particular, there are a number of published results (Rai, Felmy, and Ryan 1990, Gayer and Leider 1957, Ryan and Rai 1983, Tremain et al. 1981, Casas et al. 1998) that suggest amphotericity for U4+ at pH >10. This, however, likely resulted from combined effects of two experimental artifacts: (1) oxidation to UO22+, which is much more soluble, and (2) the presence of carbonate, which is a strong complexant of U4+.
The solubility of U(IV) phases were also determined in simplified brines under conditions that relate to the WIPP (Rai et al. 1997, Rai et al. 1998; Yajima, Kawamura, and Ueta 1995, Torerro et al. 1994). These data are shown in Figure SOTERM-9. Rai et al. (1997) determine the solubility of freshly precipitated UO2×xH2O(am) in NaCl and MgCl2 solutions of various ionic strengths. They estimate the concentration of U(OH)4(aq) in equilibrium with UO2×xH2O(am) to be about 10-8.0 M, and a number of data with greater concentrations in the neutral and alkaline range are ascribed to the presence of U(VI) in solution. This is in fair agreement with the value of 10-(8.7 ± 0.4) M proposed by Yajima, Kawamura, and Ueta (1995). It is important to note that U(IV) concentrations at pH >5 show no significant dependence on the initial solid phase (Figure SOTERM-9); both fresh precipitates in oversaturation experiments or electrodeposited microcrystalline UO2(s) in undersaturation experiments gave the same results (Torrero et al. 1994).
Figure SOTERM-8. Predominance Diagrams for U as a Function of Log PO2 and pH: (A) Predominant Solid Phases; (B) Predominant Aqueous Species. Experimental Log PO2 – pH Measurements of the Experiments from Casas et al. Open Dots: 0.008 M Perchlorate; Filled Dots: 1 M Chloride; Open Squares: Cigar Lake; Filled Squares: Jachymov; Stars: Oklo (Based on Data in Casas et al. 1998).
Figure SOTERM-9. Solubility of UO2(s) as a Function of pH at 20–25 ºC (68–77 °F) in 1M NaCl (based on Neck and Kim 2001). The Experimental Data are from Ryan and Rai (1983), Rai et al. (1997), and Neck and Kim (2001). The Solid Line is Calculated by Neck with Log Ksp = (-54.5 ± 1.0) and the Hydrolysis Constants Selected in Neck and Kim (2001). The Dotted Lines Show the Range of Uncertainty. The Dashed Line is Calculated With the Model Proposed by Rai et al. (1997).
U(VI) phases and aqueous species, although not expected to predominate in the WIPP, could be present due to the localized effects of radiolysis (see Section SOTERM-2.4.2). The WIPP PA currently makes the conservative assumption that U(VI) species predominate in 50% of the PA vectors. The solubility of U(VI) is, however, not explicitly calculated in WIPP PA, since there is no model for actinides in the VI oxidation state. The potential contribution of U(VI) species to the overall solubility of U in the WIPP is implicitly considered in the WIPP PA in the 1 mM value for U solubility (U.S. Environmental Protection Agency 2005). Prior to this, the solubility of U was defined as 1.2 × 10-5 M based on an assessment of the literature and existing WIPP-relevant experimental data by Hobart and Moore (1996).
The role of carbonate
(CO32-) in the U(VI) solubility is indeed
important (Clark, Hobart, and Neu 1995, Guillaumont et al. 2003). In the absence of competing
complexing ligands, carbonate complexation will dominate the
speciation of the uranyl ion under near-neutral pH conditions as
long as there is ample carbonate-bicarbonate available.
Complexation constants for binary U(VI) carbonate complexes at I
= 0 M and 25 ºC (77 °F) are listed in Table SOTERM-10
Table SOTERM-10. Complexation Constants for Binary U(VI) Carbonate Complexes at I = 0 M and 25 ºC (Guillaumont et al. 2003)
|
Reaction and Solubility Product for UO2CO3(crystalline [cr]) |
|
|
UO2CO3(cr) ⇌ UO22+ + CO32- |
Log K0SP(cr)= -14.76 ± 0.02 |
|
Reactions and Formation Constants β0nq for (UO2)n(CO3) q2n-2q |
|
|
UO22+ + CO32- ⇌ UO2CO3(aq) |
Log β011 = 9.94 ± 0.03 |
|
UO22+ + 2 CO32- ⇌ UO2(CO3)2 2- |
Log β012 = 16.61 ± 0.09 |
|
UO22+ + 3 CO32- ⇌ UO2(CO3)3 4- |
Log β013 = 21.84 ± 0.04 |
|
3 UO22+ + 6 CO32- ⇌ (UO2)3(CO3) 66- |
Log β036 = 55.6 ± 0.5 |
(Guillaumont et al. 2003). The three monomeric complexes of general formula UO2(CO3), UO2(CO3)22-, and UO2(CO3)34- are present under the appropriate conditions. There is also evidence from electrochemical, solubility, and spectroscopy data that support the existence of (UO2)3(CO3)66-, (UO2)2(CO3)(OH)3-, and (UO2)11(CO3)6(OH) 122- polynuclear species, which can only form under the conditions of high-metal-ion concentration or high ionic strength (Clark, Hobart, and Neu 1995). At uranyl concentrations above 10-3 M, the trimeric cluster (UO2)3(CO3)66- can also be present in significant concentrations. When the uranyl ion concentration begins to exceed the carbonate concentration, hydrolysis will play an increasingly important role (Clark, Hobart, and Neu 1995).
It is generally accepted that the major complex in solution at high carbonate concentrations is UO2(CO3)34- (Kramer-Schnabel et al. 1992, Peper et al. 2004). However, at I = 0.5 M and I = 3 M, the polynuclear (UO2)3(CO3)66- species becomes an important competitor of UO2(CO3)34-. Grenthe et al. (1984) indicated that the formation of (UO2)3(CO3)66- is favored at high ionic strengths as a result of possible stabilization of the complex by ions of the background electrolyte.
The solubility of U(VI) in the WIPP is defined by the combined contribution of two processes: hydrolysis with oxyhydroxide phase formation, and carbonate complexation with U carbonate phase formation. These are both very complex systems, and there are many proposed speciation schemes. In carbonate-free or low-carbonate solutions, the speciation of U(VI) is dominated by hydrolysis.
The solubility of uranyl carbonate UO2CO3 and the formation constants for the associated complexes were determined at 25 ºC (77 °F) in 0.1M NaClO4 by Kramer-Schnabel et al. (1992). The authors noticed a change in the composition of the solid when the pH increased above 6.5. Using 14C-labeled carbonate and X-ray analysis, they observed that UO2CO3 changed to a mixed uranyl-hydroxy-carbonate at pH >6.5 and to uranyl hydroxide or sodium diuranate at pH >8. The different transition states were not characterized in detail. In an earlier investigation, the existence of hydroxycarbonato uranyl species in the neutral pH range at 25 ºC was also determined. An important observation from these studies is that the U concentration in solutions decreases with increasing ionic strength when UO2CO3 is the major aqueous U species.
At high pH, Yamamura et al. (1998) demonstrate that hydrolysis overwhelms carbonate complexation. The solubility of U(VI) was measured in highly basic solutions (11≤ pH ≤ 14) at an ionic strength of I = 0.5 – 2 M over a wide range of carbonate concentrations (10-3 – 0.5 M) using both oversaturation and undersaturation approaches. In the oversaturation experiments, the solubility of U(VI) decreased with increasing equilibration time from one week to one year and was explained as an increase in the crystallinity of the solid phase with aging. The solid phase was identified as Na2U2O7×xH2O by XRD. The undersaturation experiments conducted for one month with the solid phase indicated a rapid equilibrium. These data were interpreted by considering the formation of UO2(OH)3-, UO2(OH)42-, and UO2(CO3)34- (Yamamura et al. 1998).
The influence of carbonate on U(VI) solubility in highly saline solutions was investigated by Lin et al. (1998) and Fanghänel and Neck (2002). Lin et al. (1998) evaluated U(VI) solubilities with up to 5M NaCl in a range of carbonate concentrations. At carbonate-ion concentrations greater than 10-7 M, UO2(CO3)34- was the dominant U(VI) complex in solution. At higher CO2 partial pressures, the solubility-controlling solid phase was found to be UO2CO3(s), whereas at lower partial pressures, sodium uranate was identified as the solid phase in NaCl-saturated solutions. This study, although interesting, is of questionable use to the WIPP because the details were not fully published.
The solubility-controlling U(VI) solid phases are schoepite-type phases UO3×xH2O(s). This was reported by Sandino and Grambow (1994) as corrosion products of spent fuel in long-term leaching experiments under oxidizing conditions. However, in complex systems where many other elements are present, the uranyl hydroxides are not predicted to predominate in the long term. Specifically, they undergo a transformation into different phases that can include divalent cations (e.g., Ca2+, Pb2+, Ba2+) and monovalent cations, such as K+ or Na+ (Sandino and Grambow 1994). The transformation reaction is generally dependent on pH and groundwater composition. As an example, Fanghänel and Neck (2002) observed the formation of a sodium (Na) uranate phase [clarkeite, NaUO2O(OH)×H2O] in 5M NaCl experiments at pH 8. The extent to which these alkali uranate salts contribute to U solubility in the higher-complexity brines expected in the WIPP is not clear.
The U solid phases formed in the presence of carbonate were also investigated. Meinrath and Kimura studied solid-liquid equilibria of U(VI) at 100, 0.98 and 0.03% CO2 partial pressures in the pH range 2.8-4.6 in 0.1M NaClO4 solution at (24 ± 2) ºC (Meinrath and Kimura 1993). The solid phase formed under 100% CO2 partial pressure was found as a faint, yellow-greenish powder. It was identified by XRD as rutherfordine (UO2CO3). At 0.98 and 0.03% CO2 partial pressures, the solids generated were bright yellow, and identified by XRD as schoepite (UO3×2H2O). The authors established that the phase transition between these two phases (rutherfordine and schoepite) occurs at a CO2 partial pressure of 2.8% (Meinrath and Kimura 1993). This estimated value is in agreement with the experimental results of Grenthe et al. (1984) who report rutherfordine as a solubility-limiting solid phase at partial pressures ≥ 4.8% CO2.
The solubility of U(VI) in the absence of carbonate was extensively studied since CRA-2004 in simulated GWB and ERDA-6 brine (Lucchini et al. 2009). An overview of these results is shown in Figure SOTERM-10, and a comparison of these results with other solubility data in the literature is given in Table SOTERM-11. The measured U(VI) solubilities were about 10-6 M in GWB brine at pCH+ ≥ 7 and about 10-8 - 10-7 M in ERDA-6 at pCH+ ≥8. These results put an upper bound of ~10-5 M for the solubility of uranyl in the carbonate-free WIPP brines for the investigated range of experimental conditions. At the expected pCH+ in the WIPP (~8.7), the measured uranium solubility was between 10-7 M and 10-6 M.
Figure SOTERM-10. U(VI) Solubility in Carbonate-Free Brine Versus pCH+ for GWB (Top Curve) and ERDA-6 (Bottom Curve). These Correspond to Data Obtained After 705-day Equilibration Using an Oversaturation Approach (Lucchini et al. 2009).
Table SOTERM-11. Solubility of U(VI) in High-Ionic-Strength Media
|
U(VI) Concentration (M) |
pCH+ |
Solution |
Time (days) |
Solid |
Reference |
|
(2.8 ± 1.8) ×10-5 |
8.9 |
5M NaCl |
≈ 50 |
Na0.68UO3.34× (2.15±0.10)H2O |
|
|
(8.2 ± 4.6) ×10-5 |
7.6 |
5M NaCl |
≈ 110 |
Na0.45UO3.23× (4.5±0.1)H2O |
Diaz-Arocas and Grambow 1998 |
|
(4.2 ±1.9) ×10-4 |
7.1 |
5M NaCl |
≈ 170 |
Na0.29UO3.15× (2.9±0.2)H2O |
Diaz-Arocas and Grambow 1998 |
|
(2.8 ± 0.9) ×10-6 |
6.5 |
5M NaCl |
≈ 170 |
Na0.14UO3.07× (2.5±0.1)H2O |
Diaz-Arocas and Grambow 1998 |
|
(1.82 ± 0.01) ×10-3 |
8.4 |
Brine |
100 |
α-schoepite (oversaturation) |
|
|
(1.81 ± 0.01) ×10-3 |
8.4 |
Brine |
100 |
α-schoepite (oversaturation) |
Yamazaki et al. 1992 |
|
(1.40 ± 0.05) ×10-3 |
8.4 |
Brine |
244 |
α-schoepite (undersaturation) |
Yamazaki et al. 1992 |
|
(1.80 ± 0.05) ×10-3 |
8.4 |
Brine |
244 |
α-schoepite (undersaturation) |
Yamazaki et al. 1992 |
|
(3.8 ± 0.4) ×10-7 |
10.4 |
Brine |
150 |
Mg(OH)2 and K2U2O7 (oversaturation) |
Yamazaki et al. 1992 |
|
(3.1 ± 0.3) ×10-7 |
10.4 |
Brine |
150 |
Mg(OH)2 and K2U2O7 (oversaturation) |
Yamazaki et al. 1992 |
|
(1.7 ±1.4) ×10-7 |
8.1 |
ERDA-6 |
705 |
To be determined (oversaturation) |
|
|
(9.9 ± 3.0) ×10-8 |
9.6 |
ERDA-6 |
705 |
To be determined (oversaturation) |
Lucchini et al. 2009 |
|
(3.1 ± 1.3) ×10-8 |
10.5 |
ERDA-6 |
705 |
To be determined (oversaturation) |
Lucchini et al. 2009 |
The most important observations from these U(VI) solubility studies were
· The measured solubility of U(VI) in the absence of carbonate was ~10 – 100 times lower that those reported by others in the literature and well below the current 1 mM limit used in the WIPP PA (which applies to all conditions, including carbonate). These lower solubilities reflect the lack of oxygen and carbonate in the brine systems investigated.
· U(VI) solubility does not exhibit amphoteric behavior.
· A difference in U(VI) solubility between the two brines investigated was noted, with U concentrations in GWB ~10 times higher at pH > 8. This is caused by the complexation of U(VI) with the higher borate and sulfate concentrations in GWB.
The new WIPP-specific data do not address the more important issue of the effects of carbonate complexation on U(VI) solubility, but they do establish a baseline to determine this effect.
The WIPP repository is projected to contain ~10 kg of Np, primarily as the 237Np isotope (see Table SOTERM-8). Its inventory increases with time from the decay of 241Am and the possibility of 238U (n, 2n) reactions, but this increase is projected to be less than a factor of two during the regulatory period of the WIPP. In the WIPP PA, Np speciates as Np(IV) in 50% of the PA vectors and as Np(V) in the other 50% of the PA vectors. The contribution of Np to actinide release from the WIPP was included in the CRA-2004 PABC (Brush and Xiong 2005a; Leigh et al. 2005) calculation, but its effect on release was negligible. Arguments have also been made that it should be excluded from consideration in the WIPP PA based on its low inventory (Brush and Garner 2005).
The environmental chemistry of Np is somewhat unique in the actinide series as a result of the relatively high stability of the NpO2+ species, which is in the V oxidation state, under a wide range of conditions typically found in the subsurface. This oxidation state is prevalent when oxidizing conditions predominate (Hobart 1990). It is mobile because it has a relatively high solubility and it is not strongly sorbed or complexed. It does not hydrolyze strongly, with little or no measurable hydrolysis until pH >7 (Neck, Kim, and Kanellakopulos 1992; Itagaki et al. 1992). Much of the complexation data for inorganic and organic complexes for Np pertains to the V oxidation state for this reason (Lemire et al. 2001). The log Ksp for NpO2OH (s) is 4.5 ± 0.06 (Neck, Kim, and Kanellakopulos 1992).
Np can, however, actually exist in up to five oxidation states in aqueous media. The redox potentials under basic conditions are (Marinot and Fuger 1985):
+ 0.58
V
+ 0.6
V
+
0.3V
-2.1V
NpO53-
→
NpO2(OH)2
→
NpO2OH
→ NpO2
→ Np(OH)3
(SOTERM.27)
Only the Np(IV) and Np(VI) oxidation states, in addition to Np(V), can exist under the right conditions in reducing or oxidizing groundwater (Hobart 1990; Keller 1971 [pp. 195–215]; Clark, Hobart, and Neu 1995). These exist as Np4+ complexes and NpO22+ complexes. Np(VI), unlike Np(V), is strongly hydrolyzed at near-neutral pH and is readily reduced by many constituents typically found in groundwater (e.g., organics and most reduced metals). For these reasons, it does not tend to persist in groundwater under most conditions.
Under reducing anoxic conditions, Np4+ species can predominate. These Np4+ species readily undergo hydrolysis and are comparable to Pu4+ in this regard. This system is highly irreversible and probably polymeric in nature, as is observed for Pu4+. The measured solubility of Np4+ is 10-8.5 to 10-8.1 M with Np(OH)4, not Np(OH)5-, as the predominant aqueous species (Rai and Ryan 1985, and Eriksen et al. 1993). The importance and predominance of the Np(IV) oxidation state in reducing conditions is even more pronounced when anaerobic bacteria are present. Np(V) was readily reduced by sulfate-reducing bacteria (Banaszak, Reed, and Rittmann 1998) and methanogenic consortia (Banaszak et al. 1999), and precipitated as Np(IV) solids.
In WIPP-specific experiments, Reed and Wygmans (1997) found spectroscopic evidence for reduction of Np(VI) to Np(V) in ERDA-6 (Castile) brine at pH 10, and have observed complete reduction of Np(VI) to Np(V) in G-Seep (Salado) brine at pH 7 when no iron or microbial activity were present. In the presence of oxalate, citrate, and EDTA, Reed and Wygmans (1997) have observed rapid and complete reduction of Np(VI) to Np(V) coupled with a slower formation of Np(IV) species. The stability of Np(V) under these conditions is further confirmed by Neck, Runde, and Kim (1995), who showed that Np(V) carbonate complexes are stable in 5M NaCl.
In the expected WIPP environment, however, where anoxic and reducing conditions with microbial activity and reduced iron are expected to be present, Np(IV) is expected to be the predominant oxidation state (Rai and Ryan 1985, Rai, Strickert, and McVay 1982a, Kim et al. 1985, Pryke and Rees 1986). This is based on studies of the solubility of NpO2OH in 1 M and 5 M NaCl solutions at pH 6.5 where the reduction of Np(V) to Np(IV) was observed (Kim et al. 1985, Neck, Kim, and Kanellakopulos 1992).
There are no new WIPP-relevant results on the chemistry and speciation of Np since CRA-2004 and the CRA-2004 PABC.
Pu is a key TRU component that contributes significantly to the potential for TRU release from the WIPP under all release mechanisms considered by PA. Pu isotopes, estimated to be ~10 metric tons at the time of closure, represent approximately 89% of the Ci content for actinides in TRU waste (see Table SOTERM-7). There are five isotopes of Pu that make a significant contribution to the Pu inventory, but 239Pu, 238Pu, and 241Pu are the major contributors to the Ci content. Under the conditions expected in the WIPP, Pu(IV) is expected to be the predominant oxidation state (Weiner 1996). A more extensive review of Pu subsurface speciation issues as they pertain to the WIPP case was completed (Reed et al. 2009).
In the WIPP PA, all of the Pu is assumed to be reduced and present in the III or IV oxidation state. Half of the PA vectors contain 100% Pu(III), with the other half of the vectors containing 100% Pu(IV) species. Because the solubility of Pu(III) is roughly 10 times higher, the assumption that it is present is a conservatism built into the WIPP PA. The two higher-valent Pu oxidation states, Pu(V) and Pu(VI), are not considered in the PA because they cannot persist under the expected reducing and anoxic conditions in the WIPP.
Generally, Pu can exist in oxidation states III, IV, V, VI, and VII (Katz, Seaborg, and Morss 1986, p. 781). Of these, only Pu(V), Pu(IV), and Pu(III) are expected to be important under environmentally relevant oxidizing and reducing conditions. Pu(VII) is very unstable and exists only in extremely basic solutions (for example, 7 M NaOH) that are not expected in the WIPP. Pu(VI) can persist in the WIPP in the absence of reductants, but is readily reduced in the presence of Fe(II/0) species, reduced by many organic chelators (Reed et al. 1998), and reduced in anaerobic, biologically active systems (Reed et al. 2007; Icopini, Boukhalfa, and Neu 2007). The reduction of Pu(VI), under WIPP-relevant conditions, was shown by Clark and Tait (1996), Reed and Wygmans (1997), and Reed et al. (2007).
The role and importance of redox reactions in determining actinide mobility and solubility are beyond question (Van Luik et al. 1987; Allard 1982; Choppin and Rao 1992). The redox potentials for the various oxidation states at pH 7 are (Cleveland 1979, pp. 11–46)
+0.77 V
+1.11
V
-0.63 V
PuO2(OH)2 (aq)
→ PuO2+
→
PuO2∙H2O (s)
→
Pu3+
(SOTERM.28)
└──────────────────┘
+ 0.94 V
A typical phase diagram for Pu in groundwater that illustrates the importance of redox is shown in Figure SOTERM-11.
Higher-valent Pu, specifically Pu(V) and Pu(VI), can be present in near-surface oxidizing groundwaters (Orlandini, Penrose, and Nelson 1986). The association of Pu(V) with organic colloidal material was proposed as the mechanism by which subsurface migration occurred. Pu(VI), in near-neutral systems, is strongly and irreversibly hydrolyzed (Okajima and Reed 1993). It is also readily reduced by organics and reduced metal species even when oxygen is present to form Pu(V), and is not generally stable under most groundwater-relevant conditions.
Pu(V), by analogy with Np(V), does not undergo hydrolysis until pH >7 and tends to form weak complexes. It readily disproportionates to form Pu(IV) and Pu(VI) at high concentrations and is relatively easy to reduce in the environment under anoxic conditions. Fe2+(aq), Fe(II) minerals, and metallic iron reduce Pu(V) to Pu(IV).
In geochemical systems, redox control is often interpreted in terms of the iron, and in a broader sense, reduced metal, mineralogy, and associated aqueous chemistry (Sanchez, Murray, and Sibley 1985, White, Yee, and Flexser 1985). In the WIPP case, iron will undergo anoxic corrosion, producing Fe2+. Both metallic iron (Fe0) and Fe2+ have been shown to quantitatively reduce Pu(VI) in the WIPP brines to either Pu(IV) or Pu(III). Clark and Tait (1996) and Felmy et al. (1996) have experimentally observed the reduction of Pu(VI) carbonates by either Fe0 or Fe2+ to Pu(IV). In the absence of carbonates, a quantitative reduction of Pu(VI) is also observed, but the oxidation state of the resulting species cannot be definitively determined because its
Figure SOTERM-11. Speciation Diagram for Plutonium in Carbonated Low-Ionic-Strength Groundwater (Based on Data Presented in Runde et al. 2002). This Illustrates the Expected Lower Solubility of Reduced Pu, Shows Pu(IV) Rather Than Pu(III) Species at Near-Neutral pH, and Suggests That the Dominant Pu Species in the pH 8-9 Range are Hydrolytic Species with Lesser Contributions From Carbonate.
concentration is below the lower detection limit of the oxidation state analytical process (about 10-9 M). However, since this concentration is well below the expected solubility of Pu(V) species, it was reasonably assumed that the Pu must have been reduced to either the IV or III oxidation state. Neretnieks (1982) has shown that when dissolved actinides in moving groundwater came in contact with Fe(II), the actinides were reduced to a much-less-soluble oxidation state and precipitated.
Pu(III) is not predicted to be stable under the expected WIPP conditions. There are some mechanisms, however, identified in which Pu(III) species can be formed. Felmy et al. (1989) observed some Pu(III) in the WIPP brines at neutral and slightly basic conditions. PA conservatively takes account of these minor mechanisms by assuming that Pu is speciated as Pu(III) in 50% of the PA vectors.
Comprehensive and critical reviews of how actinide species and microorganisms interact have been published (Banaszak, Rittmann, and Reed 1998; Neu, Ruggiero, and Francis 2002). General aspects of this were discussed in Section SOTERM-2.4.1.2. Additionally, the important role of microbial activity through biotic transformations (Francis 1990; Zitomer and Speece 1993; Banaszak, Rittmann, and Reed 1998; Rittmann, Banaszak, and Reed 2002; Reed et al. 2007) in defining oxidation state distribution of multivalent metals and actinides has been recognized.
Although the bioreduction of uranyl and neptunyl species is well established, there are relatively few studies of the bioreduction of plutonyl species. Reed et al. (2007) demonstrate that Shewanella alga, a ubiquitous metal-reducing soil bacteria, reduces Pu(V) to Pu(III/IV) species. Icopini, Boukhalfa, and Neu (2007) have shown that Geobacter and Shewanella odeinensis also reduce higher-valent Pu to Pu(III/IV) species.
These Pu data are consistent with the oxidation state predictions in microbially active systems shown in Figure SOTERM-2. It is particularly important to note that Pu(IV) is the expected oxidation state under a wide range of anoxic subsurface conditions, with no Pu(V) or Pu(VI) species expected. The recent Pu bioreduction results confirm that highly reducing conditions are being generated by metal-reducing bacteria under anaerobic growth conditions and support the current WIPP PA assumption that higher-valent actinides cannot persist when the concentration of dissolved actinides is important and microbial activity is prevalent.
There are no studies on the bioreduction of Pu(V/VI) under WIPP-relevant conditions. Halophiles (Gillow et al. 2000) typically found and expected to predominate in the WIPP environment have not been studied in the context of their tendency and ability to reduce higher-valent actinides. Since there is a high expectation that geochemical reactions alone will produce an anoxic, strongly reducing environment in the WIPP, halophiles, by analogy, are also expected to cause the bioreduction of multivalent actinides in the WIPP by both indirect cometabolic and direct enzymatic pathways.
It has long been held that Pu oxide, as PuO2, is the thermodynamically favored form of Pu oxide. This oxide is likely the predominant form of Pu in TRU waste and is believed to be the most important phase under WIPP-related conditions. In the last few years, however, there have been a number of studies that question this key and fundamental assumption.
Haschke, Allen, and Morales (2000) report that near-stoichiometric plutonium dioxide reacts with water vapor at temperatures between 25 °C and 350 °C (77 °F and 662 °F) according to the following reaction:
PuO2(s) + xH2O(g) ® PuO2+x(s) + xH2(g) (SOTERM.29)
Here, water vapor is reduced by polycrystalline PuO2 to produce hydrogen (H) and a previously unknown higher-oxide PuO2+x with x as large as 0.27. If only Pu(IV) and Pu(V) are present in PuO2.27, this oxide has 46% Pu(IV) and 54% Pu(V). Once formed, the PuO2+x may dissolve in contact with groundwater to form aqueous PuO2+ or PuO22+ species (Haschke and Ricketts 1995).
There remains some controversy about the mechanisms that led to the observation of higher-valent Pu in the PuO2+x. This process only occurs under unsaturated conditions at high relative humidities. Haschke, Allen, and Morales (2000) argue that this conversion is due to a chemical reaction (that is, the above reaction has a Gibbs energy less than zero) rather than a radiolysis-induced reaction because the reaction rate is temperature dependent. However, there seems to be some contribution from radiolysis in this process and this may be the dominant mechanism (LaVerne and Tandon 2002). Neither of these mechanisms are expected to impact WIPP repository performance.
The behavior of PuO2 in contact with water was studied as a function of time by means of the short-lived isotope 238Pu, as well as the longer-lived 239Pu (Rai and Ryan 1982b). This study concluded that crystalline PuO2, amorphous PuO2, and amorphous PuO3×xH2O all convert to a material intermediate between crystalline PuO2 and a hydrated amorphous material that contains both Pu(IV) and Pu(VI). These authors hypothesized that alpha particles generated by 238Pu or 239Pu irradiated water to generate OH radicals that reacted to form Pu(V) and/or Pu(VI) on the oxide surface. These observations are why the formation of localized oxidizing zones, where some higher-valent Pu can persist, is recognized in the WIPP PA. Reduction of these species, however, leads to a reformation of Pu(IV) hydrous oxide precipitates.
The overall issue of a thermodynamic driver for higher-valent Pu oxides, although it has received much recent attention in the literature, is not yet resolved, but has a relatively insignificant impact on the WIPP regardless of the mechanisms at work. A prolonged unsaturated phase in the WIPP could lead to the formation of some PuO2+x, but this will be quickly overwhelmed in an aqueous environment and the higher-valent Pu will be reduced to Pu(III/IV) species, as described in Section SOTERM-3.5.1.1 and Section SOTERM-3.5.1.2. Both DBR and transport-release scenarios assume brine inundation and, correspondingly, the rapid introduction of reducing conditions.
General studies of Pu in brine have been done by a number of investigators (Büppelmann et al. 1986; Büppelmann, Kim, and Lierse 1988; Clark, Hobart, and Neu 1995; Nitsche et al. 1992; Nitsche et al. 1994; Pashalidis et al. 1993; Villareal, Bergquist, and Leonard 2001; Reed et al. 1993; Reed, Okajima, and Richmann 1994; Reed and Wygmans 1997). There has also been an assessment of the actinide chemistry in the WIPP CCA (Oversby 2000; Brush, Moore, and Wall 2001; U.S. Environmental Protection Agency 2006). These studies confirm reduction of higher-valent Pu under the expected WIPP conditions and establish the key speciation trends for Pu in the WIPP. These trends, however, are captured in the WIPP PA through analogy with Am(III) for Pu(III) and with Th(IV) for Pu(IV).
WIPP-specific experiments and progress were made in two important areas since CRA-2004 and CRA-2004 PABC. The first is that a series of experiments to determine the solubility of neodymium (Nd) in the WIPP brine were completed (Borkowski et al. 2008). Nd is an oxidation-state-invariant analog for the III actinides and, in this context, is an analog for the solubility of Pu(III). These results are summarized in Section SOTERM-3.6.2 and support the current WIPP PA solubilities in the CRA-2004 PABC.
The second area of progress was in the completion and publication of WIPP-specific experiments that establish the reduction of higher-valent Pu(V/VI) species by reduced iron in the brine. A series of WIPP-specific experiments performed by researchers at Argonne were confirmed and published (Reed et al. 2006). Additionally, experiments were performed to further confirm the reduction of higher-valent Pu and establish its reactivity with iron oxides (Reed et al. 2009).
Iron and lead (Pb) have an important role as reducing agents for Pu in the V and VI oxidation state. Under the conditions expected in the WIPP, iron can exist as Fe(0), Fe(II), and small amounts of Fe(III) species, and lead can exist as Pb(0) and Pb(II) species. The expected importance of these two metals is based on the redox-half-reaction potentials for the reduced metal oxidation states Fe(0/II) and Pb(0/II) relative to Pu(V/VI), and the significant amount of these two metals present in the WIPP. Whereas, for U, the existence of favorable redox potentials are somewhat dependent on the speciation of Fe and Pb, the existence of reduced iron and Pb always lead to favorable redox conditions for the reduction of both Pu(V) and Pu(VI) species under a wide range of conditions.
Two key figures from Reed et al. (2009) that demonstrate the reduction of Pu(V/VI) are shown (Figure SOTERM-12 and Figure SOTERM-13). In Figure SOTERM-12, both powder and coupon forms of zero-valent iron led to the rapid (few days) reduction of Pu(V/VI). XANES analysis confirmed that Pu(IV) was produced.
Figure SOTERM-12. Comparison of the Reactivity of Iron Powder and Iron Coupon Towards Pu(VI). Rapid Reduction/Removal from Solution was Observed at pH 7 (GWB brine) and pH 8 (ERDA-6 brine). This was Somewhat Slower at pH 10 in ERDA-6 Brine (Reed et al. 2009).
In Figure SOTERM-13, the effect of Fe(II) in the iron phases is shown. Overall, the reduction of Pu(V/VI) is observed over a wide range of conditions in the WIPP brine when either zero-valent iron, aqueous Fe2+, or Fe(II) phases are present in the WIPP brine. When Fe(III) phases are present, only sorption, not reduction, is observed. These data provide strong and WIPP-specific evidence that reduced iron phases will reduce higher-valent Pu to Pu(IV) and support the current WIPP PA assumptions on oxidation state distribution.
Figure SOTERM-13. The Concentration of Pu as a Function of Time in the Presence of Iron Powder, Iron Coupon, Ferric Oxide, and Magnetite (Mixed Iron Oxide) (Reed et al. 2009)
There are relatively small quantities of Am in TRU waste (see Table SOTERM-8), but the high activity of 241Am (t½ = 432 years, 3.443 Ci/g) make Am a key contributor to potential actinide release from the WIPP. In the WIPP PA, Am is in the trivalent state in all vectors and the aqueous concentration consists of Am3+ complexes and colloidal species.
Cm is also present in very small quantities in the WIPP (Table SOTERM-8) and exists primarily as the 244Cm isotope. The high activity of this isotope (t½ = 18.11 years) makes Cm an important species in the WIPP at only the very early stages of repository history. It is essentially unimportant for the PA because it has decayed away by the end of the 100-year period for active institutional controls. However, other Cm isotopes with longer half-lives are present in the inventory and are considered by the WIPP PA. The environmental chemistry of Am and Cm are very similar, and most of what is said in this section about the environmental chemistry of Am also applies to Cm.
A more detailed review of the literature for Am can be found as part of a recent WIPP report (Borkowski et al. 2008). The solubility of An(III) was measured in the WIPP brine over a wide range of conditions using Nd(III) as a redox-invariant analog. These data support current WIPP PA calculations for the solubility of Pu(III) and Am(III) in the WIPP brine and are also summarized in Borkowski et al. (2008).
Am is a 5f electron element and, like other
elements of the actinide group, can exist in aqueous solution in
several oxidation states. The electrode potentials for some Am
couples are presented in Figure SOTERM-14. The trivalent state of Am is the most
stable aqueous oxidation state
Figure SOTERM-14. Redox Potential for Some Am Redox Couples (Silva et al. 1995, p. 74)
(Katz, Seaborg, and Morss 1986, p. 912), and it is quite difficult to oxidize in aqueous solution (Hobart, Samhoun, and Peterson 1982). The trivalent Am ion has an ionic radius of 97.5 picometers (pm) (coordination number [CN]=6) and its chemical properties can be used as an analog for Pu(III), which has a similar ionic radius (100 pm at CN=6) and charge density, as well as for Cm(III) (97 pm at CN=6).
The Am(II) species is italicized to stress that it is only a transient species. As discussed by Marinot and Fuger (1985), there is evidence for the formation of Am(II) in aqueous perchlorate solution in the pulse radiolysis experiment. The half-life of this species was estimated to be approximately 5 μs. This species is not observed during the electroreduction of Am(III) to the metal in noncomplexing media (David, Maslennikov, and Peretrukhin 1990).
Cm is also distinguished by the relatively great stability of the III oxidation state with respect to oxidation or reduction (Katz, Seaborg, and Morss 1986, p. 970). The stability of Cm(III) may be attributed to the half-filled f-shell electronic configuration (5f7). The oxidation of Cm(III) is achieved only with the strongest oxidizing agents, and only one report claims evidence for an oxidation state higher than IV (Korpusov, Patrusheva, and Dolidze 1975). The Cm(III) to Cm(IV) transition has not been successfully induced by ozone or electrochemically, and the Cm(IV) phosphotungstate produced by oxidizing with peroxysulfate is considerably less stable than the Am(IV) analog (Katz, Seaborg, and Morss 1986, p. 971). In the reducing environment of the WIPP repository, any higher-valent Cm produced radiolytically would be unstable. For all these reasons, the predominant oxidation state for Cm in the WIPP environment is Cm(III).
Higher-valent Am species have also been noted. Am(IV) species, with an ionic radius estimated by Shannon (1976) to be 85 pm, is only stable in the presence of strongly complexing anions such as carbonate, fluoride, phosphate, or phosphotungstate, and was never found in any appreciable amount in trivalent Am solutions.
The pentavalent and hexavalent dioxoamericium ions AmO2+ and AmO22+ can be generated under strongly oxidizing conditions. Free radicals produced from α particles in water readily reduce these dioxoamericium ions back to Am3+. In concentrated NaCl solution, in which the radiolysis products are strong oxidants, pentavalent and hexavalent Am are the predominant species (Büppelmann et al. 1986). Without an oxidant, the pentavalent dioxoamericium ion slowly disproportionates to AmO22+ and Am3+. These higher oxidation states are not stable in natural waters and can be readily reduced by action of reductants naturally present in those waters.
The speciation of Am in groundwater under mildly alkaline conditions is primarily defined by hydrolysis and carbonate complexation. Hydrolysis is generally represented by the following reaction:
Am3+ + nH2O D Am(OH)n(3-n) + nH+ (SOTERM.30)
Silva measured the 243Am(OH)3(crystalline [cr]) and Nd(OH)3(cr) solubilities in 0.1 M NaClO4 solution at 25±1 oC within the pH range 6 to 10 (Silva et al. 1995, p. 79-97). This is the only study with Am hydroxide using an x-ray-characterized crystalline solid. The solid phase was prepared by rigorously controlled, high-temperature transformation of Am(OH)3(am). Optical viewing by SEM of the solid samples at the end of the solubility experiments showed no changes in the crystal. The use of the 243Am isotope diminished α-particle damage of the crystal as a result of the 17-times-lower specific activity compared to 241Am. The weakness of this experiment was the relatively short equilibration time of only 48 days. A log (Ksp) of 16.6 ± 0.4 was obtained for the Am(OH)3 phase. The corresponding hydrolysis constants are listed in Table SOTERM-12. Similar values for Nd(III) hydrolysis were derived from the Nd(OH)3(cr) solubility measurements.
Stadler and Kim (1988) investigate the pH dependence of Am(OH)3(s) solubility in 0.1 M NaClO4 and more concentrated Na chloride and perchlorate solutions at 25 ± 0.5 oC. The effect of α-induced radiolysis on solubility was also studied using different total concentrations of 241Am. The solid phase was not characterized in this work. Although the solid used in this work was different than that used by Silva et al. (1995, pp. 275–76), the reported solubility products are in agreement. It is unclear, however, if the same phase controls the Am solubility in these two cases, because of markedly different preparation conditions of the starting solids.
Kim et al. (1984) measured the solubility of Am(OH)3(s) at I =
0.1 and 0.3 M NaClO4, in the absence of CO2
and at pCO2 =10-3.5 atm, and attributed the
solubility measured in terms of contributions from the hydroxy,
carbonato- and mixed Am hydroxy-carbonato complexes. No
characterization of the solid was reported in this work, so it
was assumed to be AmCO3OH(s). Several
investigators found that changes in the solid phase in aqueous
suspensions of Am(III) hydroxide due to aging conditions become
evident in hours and continue for weeks. Similar
Table SOTERM-12. Hydrolysis Constants of Am(III) (in Logarithmic Units) Corresponding to Equation (SOTERM.30)
|
AmOH2+ |
Am(OH)2+ |
Am(OH)3(aq) |
Medium |
Reference |
|
-7.93 ± 0.35 |
-14.77 ± 0.25 |
-24.71 ± 0.11 |
0.1 M NaClO4 |
|
|
-7.5 ± 0.3 |
-15.4 ± 0.4 |
-26.9 ± 0.5 |
0.1 M NaClO4 |
Stadler and Kim 1988 |
|
-7.8 ± 0.4 |
-15.4 ± 0.5 |
-26.9 ± 0.5 |
0.1 M NaCl |
Stadler and Kim 1988 |
|
-8.1 ± 0.3 |
-15.8 ± 0.4 |
-27.0 ± 0.5 |
0.6 M NaCl |
Stadler and Kim 1988 |
|
-7.7 ± 0.3 |
-16.7 ± 0.7 |
-25.0 ± 0.3 |
0.1 M NaClO4 |
|
|
-6.9 ± 0.2 |
|
-23.8 ± 0.9 |
0.1 M NaClO4 |
|
|
<-8.2 |
-17.1 ± 0.7 |
<-27.0 |
I → 0 |
Rai et al. 1983 |
|
-6.40 ± 0.11 |
-13.40 ± 0.16 |
-20.31 ± 0.17 |
3 M NaClO4 |
|
|
Recalculated from literature data |
||||
|
-7.0 ± 0.4 |
-15.1 ± 0.4 |
-26.4 ± 0.5 |
0.1 M NaClO4 |
|
results were reported by Felmy, Rai, and Fulton (1990). These authors measured the solubility of AmCO3OH(cr) at pCO2 =10-3 atm. The change in total Am concentration measured in this work as a function of pH was similar to that reported by Kim et al. (1984). Similar plots for the solubility of Nd in 5 M NaCl were measured by Borkowski et al. (2008); however, the Nd concentrations obtained for the comparable pCH+ values were two to three orders of magnitude greater as a result of the higher ionic strength present.
Am complexation by carbonate was extensively investigated by solvent extraction, spectrophotometry, electromigration, and solubility (Kim et al. 1984; Rösch et al. 1989; Felmy, Rai, and Fulton 1990; Meinrath and Kim 1991; Nitsche et al. 1995; Torretto et al. 1995). Manydifferent soluble species have been proposed for the Am-water-carbonate system: pure carbonate, bicarbonate, and/or mixed hydroxy-carbonate complexes. Silva et al. (1995) carefully studied and reinterpreted the literature data. It is the consensus in these studies that Am(CO3)n(3-2n), with n = 1, 2 and 3, are the predominant carbonate complexes. According to Silva et al. (1995), there is no experimental evidence for the existence of a complex with n = 4 even at the highest carbonate concentrations. The report also suggests that there is no evidence for the formation of Am(III)-bicarbonate or hydroxy-carbonate complexes in solution. These data are, however, in disagreement with the more recent work done by Fanghänel and Kim (1998), which reports spectroscopic evidence for the formation of the n = 4 species.
Data reported by Kim et al. (1984) indicate that up to pCH+ = 8.0, the carbonate complexation does not affect the solubility of Am(III). For the higher pCH+, the presence of carbonate in 0.1-0.3 M NaClO4 increases solubility of Am(III) in relation to carbonate-free systems, and at pCH+ = 10 this difference is almost 4 orders of magnitude. The predominance of carbonate complexation is observed in the pCH+ range from 7.5 to 10. At higher pCH+, hydrolysis predominates over carbonate complexation.
An extensive series of experiments were performed to determine the solubility of Nd(III) as an analog for Pu(III) and Am(III) solubility in the brine (Borkowski et al. 2008). In this study, the solubility was determined in GWB and ERDA-6 brine, over a pH range of 6-12, and as a function of carbonate concentration. These solubility data extend earlier studies in simplified brines to simulated WIPP brine compositions and cover a broader range of experimental conditions.
There are a number of key results and observations from this Nd(III) solubility experimental study. The most important of these are
1. The solubility data reported for Nd(III) in WIPP-relevant brine systems support current WIPP PA calculations of An(III) solubility, in that the calculated values remain conservative for the reference WIPP conditions. This observation is, however, qualified somewhat by the observations summarized below.
2. Specific observations and results related to the Nd(III) solubility data include the following:
A. Excellent agreement with comparable literature values for Nd(III) solubility in carbonate-free, simplified 5 M NaCl brine study was obtained. This provided an external corroboration of the experimental approach for the only system investigated that can be directly compared to other non-WIPP studies.
B. Excellent agreement was obtained between the oversaturation and undersaturation experiments performed. This is a strong indicator that the solubility, rather than steady-state metastable concentrations, was being measured.
C. The solubility of Nd(III) in simulated WIPP brine was not strongly influenced by the range of carbonate concentrations considered (as high as a total concentration of 0.01 M). This is largely due to the complexation of Nd3+ by borate, already present in the WIPP brine at much higher concentrations, which masks the effects of carbonate.
D. The solubility of Nd, in the simplified and simulated brine systems considered, does not exhibit amphoteric behavior. In this context, the solubility of Nd at pCH+ >10 is mostly controlled by hydroxide concentration and decreases with increasing pCH+. A shoulder to a varying degree, however, is noted in the Nd solubility graphs for 7.5 < pCH+ < 10.5 as a result of complexation in all three brines investigated.
E. The shoulder in the Nd solubility data for ERDA-6 and GWB brine is caused by borate complexation. This establishes borate as the predominant complexant in brine in the pCH+ range of 7.5 to 10 (this includes the current reference pCH+). The formation constant for this complex was established to be log K of approximately 3 to 4.
It is important to emphasize that the measurement of Nd(III) solubility in GWB and ERDA-6 brines with carbonate showed that there was little effect of carbonate on Nd solubilities in these brines. This was due to the competition between borate and carbonate in these systems. Borate is, in fact, the key complexant in the WIPP brine, with its current GWB and ERDA-6 formulations, for An(III). These solubility data, however, support the current calculated III solubilities in the WIPP PA. It is the competition between borate and carbonate that makes carbonate a relatively unimportant complexant for the conditions expected in the WIPP. A composite of all literature values, including our WIPP-specific data, is shown in Figure SOTERM-15.
Figure SOTERM-15. Composite of Nd Solubility Trends Under All Conditions Investigated (Borkowski et al. 2008). Open Symbols Correspond to Undersaturation Experiments and Closed Symbols Correspond to Oversaturation Experiments.
Based on these results, there should be no significant change to the solubility of An(III) concentrations used in the WIPP PA for the reference case. In effect, although borate complexation is not currently in the model, the concentrations of III actinides calculated are conservatively high when compared to the experimental results. The WIPP-relevant data summarized in this report support current PA calculations performed with the use of the Pitzer model (U.S. Department of Energy 2004, Appendix PA, Attachment SOTERM) for the values of 3 ´ 10-7 M and 1.7 ´ 10-7 M in GWB and ERDA-6, respectively, at pCH+~8.5. The data show that this solubility is at or near the maximum solubility over a wide range of pH, brine composition, and carbonate concentrations.
The stability constants for organic
ligand-actinide complexation were determined as part of the WIPP
ASTP at Florida State University (Choppin et al. 1999). These data are summarized in Table SOTERM-13 and demonstrate some key trends in actinide
complexation. For acetate,
Table SOTERM-13. Apparent Stability Constants for the Complexation of Organic Ligands with Actinides in NaCl Media (Choppin et al. 1999)
|
Organic Ligand |
Actinide Ion |
NaCl (molality) |
log10 β1 |
|
Acetate |
Am3+ Th4+ NpO2+ UO22+ |
0.3 to 5 0.3 to 5 0.3 to 5 0.3 to 4 |
1.44 - 2.2 3.68 - 4.18 1.05 - 1.8 2.23 - 3.09 |
|
Oxalate |
Am3+ Th4+ NpO2+ UO22+ |
0.3 to 5 0.3 to 5 1.0 to 5.0 0.3 to 5 |
4.17 - 4.63 7.04 - 7.47 3.62 - 4.63 5.82 - 6.7 |
|
Citrate |
Am3+ Th4+ NpO2+ UO22+ |
0.3 to 5 0.1 to 5 0.1 to 5 0.3 to 5 |
4.84 - 5.9 9.31 - 10.18 2.39 - 2.56 7.07 - 7.32 |
|
EDTA |
Am3+ Th4+ NpO2+ UO22+ |
0.3 to 5 0.3 to 5 0.3 to 5 0.3 to 4 |
13.76 - 15.1 15.56 - 16.94 5.45 - 6.7 10.75 - 12.16 |
oxalate, and citrate, the strength of the complex formed is in the same order: IV > VI > III > V. For EDTA, the VI and III are switched. For the most part, the III and IV actinides, which are the two most important oxidation states in the WIPP, are strongly affected by organic complexation and thus can out-compete carbonate and hydrolysis if the organic concentrations are high enough. Of the four organic chelating agents considered, only citrate and EDTA are expected to form strong enough complexes to influence the speciation of actinides and potentially increase actinide concentrations under the expected conditions in the WIPP.
Actinide colloids in the WIPP are potentially important since the actinide source term is defined by the WIPP PA as the sum of contributions from dissolved actinide species and mobile colloidal actinide species (see U.S. Department of Energy 2004, SOTERM 2004) for a more detailed discussion of WIPP-relevant colloids). The importance of colloids in the migration and transport of actinide contaminants, although it continues to receive attention in the literature, remains somewhat controversial and difficult to prove. In this context, the consideration of colloidal enhancement of actinide concentrations by the WIPP PA is, at least in part, a conservatism that is built into the overall PA approach. In this context, the sorption of colloidal actinides onto fixed substrates and their filtration in low-porosity media will also reduce the mobile colloidal actinide source term, but no credit is currently being taken for this potentially significant reduction in colloidal concentrations.
Actinide colloids or pseudocolloids may be generated in the WIPP repository as a result of
1. Hydrolysis (intrinsic chemistry).
2. The interactions of dissolved actinide species with microbially derived colloids or colloids formed due to the corrosion of steel and waste constituents.
3. The hydrodynamic entrainment of colloidal-sized mineral fragments, as well as several other mechanisms. The formation of colloids could enhance actinide release in two ways. First, increased actinide concentration will increase the magnitude of DBR release and the effective actinide source term concentration for transport through the Culebra. Second, colloids have very different transport properties than dissolved species, and are predicted to migrate more rapidly in the subsurface. This transport mechanism could enhance the overall actinide release in the WIPP through migration pathways in the Culebra member and the Salado.
In this section, the general environmental aspects of colloid-enhanced transport in the subsurface are discussed, along with an update of relevant WIPP-specific results since the CRA-2004 PABC.
The potential for colloidally-enhanced transport of actinides in the subsurface continues to receive much attention in the literature. A key role of colloids in actinide transport has been proposed to explain actinide migration at Rocky Flats (LoPresti, Conradson, and Clark 2007), the Nevada Test Site (Kersting et al. 1999; Zavarin et al. 2003), Hanford (Dai, Buesseler, and Pike 2005), the Savannah River Site (Dai, Kelly, and Buesseler 2002), and the Mayak site (Novikov et al. 2006). Colloidal transport at these sites provides an explanation for subsurface actinide migration that exceeds the rates predicted for dissolved actinide species. There continues to be very weak evidence for significant transport of colloids, once formed, in natural systems.
An important theme to recent field observations of actinide colloids is the tendency of Pu, as Pu(IV), to form iron and manganese (Mn) oxide pseudocolloids. The colloidal transport of Pu in the far-field was investigated by Novikov et al. (2006) at the Mayak site in Russia. They found that the mobility of Pu in groundwater was facilitated by submicron-sized colloids. Pu(IV) hydroxides or carbonates adsorbed on amorphous iron oxide colloids were most transported. These Pu colloids were essentially removed from groundwater, leading to a drop in the Pu concentration from 1000 becquerel (Bq)/L to 0.16 Bq/L over a distance of 3 km.
The field observations are supported by laboratory studies that show a high tendency of lower-valent actinides to form iron and Mn pseudocolloids in environmentally relevant systems. Zavarin et al. (2003) shows that, at pH 8, there is a strong sorption of Pu(IV) in groundwater to birnessite (Mn-oxide) and goethite (Fe-oxide) rather than clinoptilolite (a zeolite) and calcite. Sorption was rapid and equilibrium was reached after 24 hours. Complexation with carbonate reduced Pu(IV) sorption to clinoptilolite about 15%. For iron and Mn oxides, Pu(V) sorption was also rapid, but led to the reduction of Pu(V) to Pu(IV). Khasanova et al. (Khasanova et al. 2007) also studied iron and Mn oxide interactions with actinides and saw a strong association between the dissolved actinide species and the oxides.
The potential formation of actinide pseudocolloids by association of dissolved actinides with biogenic and humic (natural) organics has also been established in the laboratory and the field. Santschi et al. (Santschi, Roberts, and Guo 2002), Asbury et al. (Asbury et al. 2001), and Orlandini, Penrose, and Nelson (1986) all show that actinides associate strongly with natural organics. These have been implicated as a potential explanation for actinide migration at Rocky Flats and in near-surface groundwater transport as a result of fallout.
Lastly, the formation of intrinsic colloids (colloids that are polymers of actinides) are important because they potentially add to the concentration of actinides in groundwater, but also because they potentially contribute to measured solubilities if care is not taken to properly account for their formation. The tendency of actinides to hydrolyze and to polymerize to form intrinsic colloids follows the order (Cleveland 1979, pp. 11–46; Choppin 1983; Kim 1991; Lieser et al. 1991)
An4+ >> AnO22+ > An3+ > AnO2+ (SOTERM.31)
The most well known and well studied actinide intrinsic colloid is the Pu(IV) intrinsic colloid, which has been used as a basis of comparison for investigating intrinsic colloids of other actinides. A discussion of colloidal Th, also in the IV oxidation state, was presented in Section SOTERM-3.2.
The most convincing and consistent explanation for the chemistry of these Pu(IV) intrinsic colloid is presented by Johnson and Toth (1978). Pu polymerization occurs nearly immediately after the first hydrolysis occurs. The following reaction involving polymerization of two hydrolyzed species by loss of H2O (olation) is proposed:
2{[(H2O)d-2Pu(OH)(H2O)](y-1)+}
D
[(H2O)d-2Pu(OH)(OH)Pu(H2O)d-2]
2(y-1)+ +
2H2O
(SOTERM.32)
Aging or maturation of the polymer then occurs by loss of H2O (olation) as follows:
[Pu(OH)(OH)Pu(OH)(OH)Pu(OH)(OH)...]n D
[Pu(O)Pu(O)Pu(O)...]n
+
3nH2O
(SOTERM.33)
An important insight into the important role of Pu polymer formation was reported by Neck et al. (2003), which investigated the solubility of Pu hydroxides/hydrous oxides under reducing conditions and in the presence of oxygen. The experimental data and thermodynamic calculations show that, under reducing conditions in the stability field of water, Pu(OH)3(s) is not stable and it converts to PuO2(s,hyd). It also found that small Pu(IV) colloids/polymers, present in neutral to alkaline solutions at the constant level of log[Pu(IV)]coll = -8.3 ± 1.0, play an important role in defining the redox potentials in these systems. The experimental results in these systems including colloid species can be described in terms of equilibrium thermodynamics. These data argue for a thermodynamically stable Pu(IV) oxidation state in the WIPP.
Lastly, there is a growing debate about the care needed in solubility studies to account for colloids in the solubilities measured—which is not a trivial problem, as the colloids are often very small (< 20 nm) and difficult to detect experimentally. The role of colloid formation, especially for An(IV) solubilities, was discussed by Fanghänel and Neck, who state, “The formation of amorphous and crystalline solids and the discrepancies between the corresponding experimental solubility data may be explained as an effort of particle size. … the predicted solubilities are often significantly lower than experimental data indicating that that solubility is controlled by the surface properties” (Fanghänel and Neck 2002). In this context, existing solubility data in the literature may include significant colloidal enhancement and overestimate the corresponding solubility.
In conclusion, there is ample evidence that colloids can form and are readily generated in the laboratory. Intrinsic colloids tend to be very low in concentration and comparable to the solubilities observed. Significant enhancement can be observed when actinides associate with oxide mineral colloids and natural and biogenic organic species. However, there remains high uncertainty in the ability of these colloidal species to migrate in the subsurface. This key issue was raised by Kersting et al. (1999) for the Nevada Test site, Dai et al. for the Hanford and Savannah River site (Dai, Kelly, and Buesseler 2002; Dai, Buessler, and Pike 2005), and strong attenuation was noted at the Mayak site (Novikov et al. 2006). In the WIPP, with its very low porosity, it is reasonable to predict that the transport of colloids is likely to be negligible; the only significant concern would be the colloidal contribution to dissolved concentrations for DBR-type release.
There are no new experiments since the CRA-2004 PABC that investigate the formation and transport of actinide colloids under WIPP-relevant conditions. Recently published results (Wall and Mathews 2005) demonstrate that the presence of MgO in the WIPP brine will significantly reduce the concentration of humic acids (HA); this occurs after a relatively short period of time (12 to 60 days) when a negligible concentration of HA was observed in the system. This important observation was attributed to MgO-facilitated HA precipitation and/or sorption of the HA onto the MgO surface. Treatment of colloids in the PA are the same as in CRA-2004 and CRA-2004 PABC.
There are no significant changes in the general approach and assumptions used to understand and predict actinide behavior in the WIPP from a PA perspective. Specifically,
· Oxidation state distributions for the TRU actinides, and correspondingly, assumptions regarding their solubility calculations using redox-invariant analogs, have not changed.
· Predicted and calculated solubilities for Pu and Am oxidation states, which are the key actinides from the perspective of PA, have not changed.
· Inventory assumptions regarding the amounts of organic chelating agents and actinides in TRU waste are being updated annually.
· The recognition that microbial colloids are the most likely to be generated in the WIPP has not changed. Treatment of colloids in PA are the same as in CRA-2004 and CRA-2004-PABC.
There are new data, within and outside the WIPP project, that continue to support and/or expand the robustness of the current PA assumptions. The most important of these are
· Extensive data from the Karlsruhe (German) program for III and IV actinides in simplified brine systems. These data support existing PA assumptions and show that they extend beyond the relatively narrow pH range considered in the WIPP PA. This is especially important for higher-pH environments, where it was previously thought that solubilities increase greatly.
· WIPP-specific results are reported in three key areas:
– An(III) solubility in simulated WIPP brines over a wide range of conditions using Nd(III) as an analog for Pu(III) and Am(III). These data support current PA solubilities for the III actinides, but show that complexation with borate explains the observed trends with pH and little or no effect of carbonate.
– The reduction of Pu(V/VI) in WIPP brine by reaction with reduced iron species. These results provide additional support to past observations that higher-valent actinides cannot persist in the WIPP in the presence of reduced iron. This strongly supports current PA assumptions on oxidation state distribution for both Am and Pu.
– The solubility of U(VI) in simulated WIPP brine over a wide range of conditions in the absence of carbonate. These data support the current WIPP PA assumptions about the solubility of U(VI).
Lastly, there are some new developments reported in the literature that, although not directly relevant to the WIPP, indirectly affect how the actinide chemistry in the WIPP is viewed. The most important of these are
· The potential role of Ca2+ and Mg2+ to form soluble species in the presence of carbonate at high pH. This has been evaluated in the WIPP case and is not likely to affect actinide solubility in the range of conditions expected in the WIPP.
· Growing recognition that microbes, under most anaerobic conditions, reduce higher-valent actinides in the subsurface.
· Additional results on the potential effects of radiolysis on brine systems. It is clear that mechanisms exist that can lead to the oxidation of actinides when no reducing agents are present in the brine. This could create localized oxidation in the WIPP, but WIPP-specific experiments show this to be easily overwhelmed by the expected microbial and reduced-iron effect on actinide redox.
The calculation of the WIPP actinide source term was performed for the CRA-2004 PABC (Brush and Xiong 2005a) using the computer code FMT. This is the baseline PA currently being used for CRA-2009. A general description of the modeling approach to establish the actinide source term for the WIPP PA is described in this section. The approach used in the CRA-2004 PABC calculations and the results obtained were published in a series of reports and documents. These are listed below with supporting letters and documentation.
Table SOTERM-14. List of Documents and Reports that Support the CRA-2004 PABC
|
PABC Analysis |
Title/Subject of Report |
|
CRA-2004 PABC (Leigh et al. 2005) |
2004 Compliance Recertification Application Performance Assessment Baseline Calculation |
|
Analysis Plan (AP)-120, Rev. 0 (Brush and Xiong 2005b) |
Calculation of Actinide Solubilities for the WIPP Performance-Assessment Baseline Calculations |
|
Letter Report: Organic ligand concentrations Task 1, AP-120, Rev. 0 (Brush and Xiong 2005a) |
Calculation of Organic-Ligand Concentrations for the WIPP PABC |
|
FMT_050405.CHEMDAT Task 2, AP-120, Rev. 0 (Xiong 2005) |
CRA-2004 PABC version of FMT thermodynamic data base |
|
Letter Report: Uncertainty Analysis Task 3, AP-120, Rev. 0 (Xiong, Nowak, and Brush 2005) |
Updated Uncertainty Analysis of Actinide Solubilities for the Response to EPA Comment C-23-16, Rev. 1 |
|
Letter Report: Actinide Solubilities Task 4, AP-120, Rev. 0 (Brush 2005) |
Results of Calculations of Actinide Solubilities for the WIPP PABC |
|
CRA-2004 PABC Inventory Document (Leigh, Trone, and Fox 2005) |
TRU Waste Inventory for the 2004 CRA PABC |
|
Actinide Concentration input to PANEL (Garner and Leigh 2005) |
Analysis Package for PANEL: CRA-2004 PABC |
|
Supporting Letter or Document |
Title/Subject of Report |
|
Sandia National Laboratories (SNL) Report (Brush et al. 2006) |
Consumption of Carbon Dioxide by Precipitation of Carbonate Minerals Resulting for Dissolution of Sulfate Minerals in the Salado Formation In Response to Microbial Sulfate Reduction in the WIPP |
|
Letter Report: Stein to Brush, 4/13/2005 (Stein 2005) |
Estimate of Volume of Brine in Repository that Leads to a Brine Release |
|
Letter Report: Brush to Kessel, 2/1/2005 (Brush and Garner 2005) |
Additional Justification for the Insignificant Effect of Np on the Long-Term Performance of the WIPP |
|
Telecon: EPA with DOE/SNL/Los Alamos National Laboratory (LANL), 3/2/2005 (U.S. Environmental Protection Agency 2005) |
Change in U(VI) Solubility Assumption to a Concentration of 1 mM |
|
Letter: Cotsworth to Triay, 3/4/2005 (Cotsworth 2005) |
Untitled: EPA documentation of requested changes to the CRA-2004 PA |
|
EPA Response and Comments on CRA-2004 PABC (U.S. Environmental Protection Agency 2006) |
Evaluation of the Compliance Recertification Actinide Source Term and Culebra Dolomite Distribution Coefficient Values |
The overall approach used to establish the actinides important in WIPP releases and calculate their solubilities for use in the WIPP PA is summarized in this section. This approach consists of the following:
· Assessing the WIPP inventory and regulations that govern the application of the WIPP certification to determine the likely actinides of interest and, correspondingly, the key waste components that may affect their solubility.
· Establishing a conceptual model for the key subsurface interactions and release mechanisms and using a combination of literature review and WIPP-specific experimental results to establish the likely oxidation state distribution, the species that affect actinide solubility, and the parameters required to model the system at high ionic strength. This approach featured the following:
– Conservative assumptions, within the bounds of the conditions expected, for the oxidation state distribution.
– Use of redox-invariant analogs for multivalent actinides to determine formation constants and establish oxidation-specific solubilities.
– Use of the Pitzer formalism and associated parameters to model solubilities at the high ionic strengths present. The Pitzer approach is recognized as the best approach for I > 0.3 M in brine systems.
· Calculating the solubility of the key actinides in the WIPP using the FMT code. The solubilities are modeled in reacted GWB and ERDA-6 brines, which are expected to bracket the range in the composition of the brine expected. This code assigns the actinides to the key species by minimizing the total free energy of the system while satisfying charge-balance and mass-balance constraints based on the standard chemical potentials assigned to each species.
· Establishing the effects of colloids and organic complexation, separately and simultaneously, on the solubilities calculated.
· Tabulating and assigning uncertainty distributions in the range of expected conditions and brine compositions to these solubility data.
This range of possible solubilities for a wide range of possible conditions defines the actinide source term provided to the WIPP PA for the calculation of TRU release from the WIPP.
The solubility and speciation of multivalent actinides are often investigated with lanthanide and actinide analogs that mimic the property of interest but, for varying reasons, provide an advantage to the experimenter. The best example of this, used extensively in the WIPP modeling approach, is the use of redox-invariant analogs for the multivalent actinides, most notably Pu, to determine oxidation-state-specific properties (e.g., solubility or complexation). The advantage of these types of analogs is that they remove the uncertainty of oxidation-state change from the experiment, which is a complexity that can often lead to uncertain or incorrect interpretations of the results obtained.
For the TRU actinides, the redox-invariant analogs used are lanthanides or other actinides. Lanthanides, as 4f-electron elements, possess physical and chemical characteristics that make them good analogs for the actinides when they are redox-invariant under the conditions of the experiment. Correspondingly, actinides with their 5f-electron character also have good physical and chemical properties to be analogs for other actinides if they also have redox stability under WIPP-relevant conditions. This analog approach, although sometimes criticized in the literature, considerably simplifies experimental design and consequently improves the reliability of the experimental data (Choppin 1999).
A key argument for the use of analogs in WIPP-related experiments is that key complexants that define actinide solubility in the WIPP are hard-donor complexants (e.g., hydroxide, carbonate, borate, chloride, and/or sulfate). The use of lanthanides as analogs for actinides is based on observations in many extraction systems, along with the associated crystallographic data (Siekierski 1988) that show they are good analogs for compounds containing hard donor ligands (oxygen) where the cation-anion interactions are primarily electrostatic in nature. In this context, Nd(III) is a good analog for the chemical behavior of Am(III) and Pu(III) under most circumstances in the WIPP. Not only do these species have the same 3+ charge, they also have similar ionic radii for coordination number 6 (CN=6): 97.5 pm for Am3+, 98.3 pm for Nd3+, and 100 pm for Pu3+ (Shannon 1976). In this context, the magnitudes of electrostatic attractions between these metal ions and corresponding ligands will be similar, yielding comparable thermodynamic stabilities.
Th is used by the WIPP as a redox-invariant analog for Pu(IV), U(IV), and Np(IV). The use of the Th4+ stability constants to represent the other An(IV) species is conservative. Th4+ is the largest of the tetravalent actinide ions. It therefore has the lowest charge density and, correspondingly, relatively weaker ionic interactions when compared to the other tetravalent actinides. This is best exhibited by its lower tendency towards hydrolysis and intrinsic polymer formation relative to the other actinides (see Section SOTERM-3.2). For these reasons, the use of Th4+ as an analog is conservative, as Th will likely be the most soluble of the actinides in the tetravalent state under comparable WIPP-relevant conditions.
To a lesser extent, actinides are analogs for each other, depending on the oxidation state. Np(V), which has much greater redox stability than Pu(V) and much more favorable spectroscopy, is often used as an analog for Pu(V). U(VI), which is much more redox stable than Pu(VI) and Np(VI), is also used as an analog for these TRU actinides, although this breaks down somewhat quickly. Am(III) and Cm(III) are also excellent analogs for Pu(III) as a result of their much greater redox stability and comparable ionic radii.
The actinide inventory used in CRA-2004 PABC is given in Table SOTERM-15 (Leigh, Trone and Fox 2005). This is based on the inventory given in Table SOTERM-7 projected to the year 2033, which is the projected year for the closure of the WIPP.
Table SOTERM-15. WIPP Radionuclide Inventory at Closure (in 2033) Used in PABC-2005 Calculations (Leigh, Trone, and Fox 2005)
|
Radionuclide |
Activity (Ci) |
Amount (kg) |
Element-Specific Inventory |
|
229Th |
5.21E+00 |
2.64E-02 |
8.81 Ci 3.11E+04 kg |
|
230Th |
1.80E-01 |
8.73E-03 |
|
|
232Th |
3.42E+00 |
3.11E+04 |
|
|
233U |
1.23E+03 |
1.27E+02 |
1.80E+03 Ci 6.47E+05 kg |
|
234U |
3.44E+02 |
5.52E+01 |
|
|
235U |
5.01E+00 |
2.32E+03 |
|
|
236U |
2.87E+00 |
4.43E+01 |
|
|
238U |
2.17E+02 |
6.44E+05 |
|
|
237Np |
1.22E+01 |
1.73E+01 |
12.1 Ci 17.3 kg |
|
238Pu |
1.13E+06 |
6.60E+01 |
2.26E+06 Ci 9.87E+03 kg |
|
239Pu |
5.82E+05 |
9.38E+03 |
|
|
240Pu |
9.54E+04 |
4.19E+02 |
|
|
241Pu |
4.48E+05 |
4.35E+00 |
|
|
242Pu |
1.27E+01 |
3.23E+00 |
|
|
244Pu |
5.53E-03 |
3.09E-01 |
|
|
241Am |
5.17E+05 |
1.51E+02 |
5.179E+05 Ci 151 kg |
|
243Am |
7.87E+01 |
3.94E-01 |
|
|
244Cm |
2.13E+03 |
2.63E-02 |
2.13E+03 Ci (0.0263 kg) |
|
137Cs (see Note a) |
2.07E+05 |
2.40E+00 |
2.07E+05 Ci (2.40 kg) |
|
90Sr (see Note a) |
1.76E+05 |
1.29E+00 |
1.76E+05 Ci (1.29 kg) |
|
a Fission products are not TRU, but are considered in the PA to calculate overall release |
|||
The oxidation states used by the WIPP PA to model actinide solubility are tabulated in Table SOTERM-16. Also included are the assumed abundance percent of each oxidation state and the speciation data set used in FMT for each oxidation state. This table is based on a general understanding of the corresponding actinide chemistry summarized in Section SOTERM-3.0.
A number of conservative assumptions are reflected in this table. The most important assumptions are
Table SOTERM-16. Oxidation States of the Actinides in the WIPP as Used in the CRA-2004 PABC
|
Actinide Element |
Oxidation States, Abundance (%), and Analog Used (If Any) |
||||
|
Oxidation Statea,b |
FMT Speciation Data Used |
||||
|
III |
IV |
V |
VI |
||
|
Thorium |
— |
100 % |
— |
— |
Thorium |
|
Uranium |
— |
50 % |
— |
50 % |
1 mM assumed for VI, |
|
Neptunium |
— |
50% |
50 % |
— |
Np for V |
|
Plutonium |
50 % |
50 % |
— |
— |
Am for III |
|
Americium |
100 % |
— |
— |
— |
Americium |
|
Curium |
100 % |
— |
— |
— |
Americium |
|
a Oxidation state distributions (percentages) refer to the percent of PA vectors that have 100% of the specified oxidation state. b In PA calculations the distribution of oxidation states is correlated for U, Np, and Pu such that the states for all three elements are simultaneously either in the lower oxidation state (U(IV), Np(IV), and Pu(III)) or in the higher oxidation state (U(VI), Np(V), and Pu(IV)). |
|||||
1. Use of 1 mM concentration for the solubility of U(VI). The actual solubility of U(VI) in the WIPP under the expected range of conditions is estimated to be <0.1 mM.
2. Use of Th as an analog for the IV actinides (see Section SOTERM-4.1 and Section SOTERM-3.2).
3. The assumption that 50% of the vectors have Pu(III) and 50% of the vectors have Pu(IV). The predominant Pu species expected is Pu(IV), although some Pu(III) is possible as a transient (see discussions in Section SOTERM-3.3). This is conservative because Pu(III) is approximately 6 to 10 times more soluble than corresponding Pu(IV) phases.
4. The assumption is that 50% of the vectors have U(IV) and 50% of the vectors have U(VI). The predominant uranium species expected is U(IV), which is approximately four orders of magnitude less soluble than U(VI), based on current assumptions.
The version of the FMT code used in the CRA-2004 PABC was FMT_050405.CHEMDAT (Xiong 2005). The data in FMT was previously described in a series of memoranda by Giambavlo (Giambavlo 2002a, 2002b, 2002c, 2002d, 2002e, 2003). The most recent database iteration included some minor changes from previous versions that go beyond those described in these memoranda:
· The chemical potential for the solubility of Th(OH)4 (s) was changed.
· The effects of hydromagnesite and calcite precipitation were added.
The thermodynamic database for the III actinides currently used in FMT was described by Giambalvo (2002a). Nd, Am, and Cm are generally used to establish solubility of An(III) because, unlike plutonium, they have redox-stable trivalent oxidation states. Speciation and solubility data for the III actinides were parameterized for use in the Pitzer activity-coefficient model by Felmy et al. (1989) for the Na+- Pu3+-Cl--H2O system; by Felmy, Rai, and Fulton (1990) for the Na+-Am3+-OH--HCO3 --H2O system; by Rai, Felmy, and Fulton (1995) for the Na+-Am3+-PO43--SO 42--H2O system; and by Rao et al. (1996) for the Na+-Nd3+-CO32--HCO 3--H2O system. For this reason, FMT uses the Am(III) data to calculate the solubility for all the III actinides. A diagram of the predominant species for Am is shown in Figure SOTERM-16.
The inorganic aqueous and solubility-limiting species featured in the model for Am(III) are
|
Am(III) Reactions |
log K |
|
|
|
Am3+ + CO32- |
8.1 |
||
|
Am3+ + 2CO32- |
13.0 |
||
|
Am3+ + 3CO32- |
15.2 |
||
|
Am3+ + 4CO32- |
13.0 |
||
|
Am3+ + OH- |
6.4 |
||
|
Am3+ + 2OH- |
12.3 |
||
|
Am3+ + 3OH- |
16.3 |
||
|
Am3+ + Cl- |
0.24 |
||
|
Am3+ + 2Cl- |
-0.74 |
||
|
Am3+ + SO42- |
3.25 |
||
|
Am3+ + 2SO42- |
3.7 |
||
|
Am3+ + OH- +
CO32- |
22.7 |
||
|
Na+ + Am3+ +
2CO32- +6H2O |
21.4 |
||
|
Am3+ + PO43- |
24.8 |
||
In these reactions, “aq,” “cr,” and “s” are the abbreviations for aqueous, crystalline, and solid, respectively. The An(III) database was extended to mixed Na+-CO32--Cl-- media, and was shown to reproduce the independently measured solubility of NaAm(CO3)2(s) in 5.6 M NaCl (Runde and Kim 1994) and the measured Nd(III) solubility in the WIPP brine (Borkowski et al. 2008).
The IV actinides addressed by the WIPP PA are
Th(IV), U(IV), Pu(IV), and Np(IV). The variation in
charge-to-radius ratio for the tetravalent actinides is greater
than for actinides in
other oxidation states (Cotton and Wilkinson 1988, pp. 11–46), and larger
differences in the chemical behavior among the IV actinides is
expected. The application of the Th(IV) model to the other
IV species is more uncertain, yet still conservative because
Th(IV) is the most soluble of these elements under WIPP
conditions. The model was evaluated against data for Pu(IV)
and Np(IV) solubility and demonstrated to predict the chemical
behavior of these actinides conservatively.
Figure SOTERM-16. Predominant Am Species as a Function of pH and Eh Based on the Speciation Reactions (SOTERM.34) to (SOTERM.47) (Richmann 2008)
The thermodynamic database for the IV actinides currently used in FMT was described by Giambalvo (2002c). Speciation and solubility data for Th(IV) were parameterized for the Pitzer activity-coefficient model for the Na+-K+ -Mg2+-Cl–- SO42--CO32--HCO3 - -OH--H2O system. This model requires the species Th4+, Th(OH)2SO4 (s), Th(SO4)32-, Th(SO4)2 (aq), ThO2, Th(OH)4(aq), Th(OH)3CO3-, and Th(CO3)56- to describe the data pertinent to the WIPP (Felmy, Mason, and Rai 1991; Rabindra et al. 1992; Felmy et al. 1996). A diagram of the predominant Th speciation, based on Reactions (SOTERM.48) to (SOTERM.59), is shown in Figure SOTERM-17.
Figure SOTERM-17. Predominant Species of Th as a Function of pH and Redox Conditions (Richmann 2008). Thorianite is Predicted to Predominate at the Conditions Expected in the WIPP Repository.
The inorganic aqueous and solubility-limiting species featured in the IV model are
|
Th(IV) Reactions |
log K |
|
|
ThO2(am) + 2H2O |
-7.0 |
|
|
Th4+ + 4OH- |
38.5 |
|
|
Th4+ + 3OH- +
CO32- |
38.3 |
|
|
Th4+ + 5CO32- |
27.1 |
|
|
Th4+ + 2SO42- |
11.6 |
|
|
Th4+ + 3SO42- |
12.4 |
|
|
Th4+ + 2SO42- +
9H2O |
13.0 |
|
|
Th4+ + 2SO42- +
8H2O |
12.9 |
|
|
Th4+ + 2Na+ +
3SO42- + 6H2O Th(SO4)2×Na2SO4×6H2O(s) |
17.6 |
|
|
Th4+ + 2K+ +
3SO42- + 4H2O Th(SO4)2×K2SO4×4H2O(s) |
18.1 |
|
|
Th4+ + 4K+ +
4SO42- + 2H2O Th(SO4)2×2K2SO4×2H2O (s) |
21.2 |
|
|
Th4+ + 7K+ +
5.5SO42- |
24.7 |
The only V actinide of interest to the WIPP is Np(V), which exists as the neptunyl ion, NpO2+. Pu(V), which can be formed under some conditions, is transitory and not expected to persist in significant quantities in the WIPP. The base model for Np(V) comes from Fanghänel, Neck, and Kim (1995), constructed for the German repository program.
The thermodynamic database for the V actinides currently used in FMT is described by Giambalvo (2002d). Np(V) speciation and solubility were parameterized in the Pitzer activity-coefficient model for the Na+-K+ -Mg2+-Cl–- SO42--CO32--HCO3 - -OH--H2O system. The model requires the aqueous species NpO2+, NpO2OH(aq), NpO2(OH)2-, NpO2CO3-, NpO2(CO3)23-, and NpO2(CO3)35-, and the solid species NpO2OH(am), NpO2OH(aged), Na3NpO2(CO3)2(s), KNpO2CO3×2H2O(s), K3NpO2(CO3)2×0.5H2O(s), and NaNpO2CO3×3.5H2O(s) to explain the available data. The predominant species for Np(V) are shown in Figure SOTERM-18.
The inorganic aqueous and solubility-limiting species used are
|
Np(V) Reactions |
log K |
|
|
|
NpO2+ + OH- |
2.7 |
||
|
NpO2+ + OH- |
8.8 |
||
|
NpO2+ + OH- |
9.5 |
||
|
NpO2+ + 2OH- |
4.5 |
||
|
NpO2+ + CO32-
|
5.0 |
||
|
NpO2+ + 2CO32-
|
6.4 |
||
|
NpO2+ + 3CO32-
|
5.3 |
||
|
Na+ + NpO2+ + CO32- + 3.5H2O ⇌ NaNpO2(CO3)×3.5H2O(s) |
11.1 |
|
|
|
3Na+ + NpO2+ + 2CO32-⇌ Na3NpO2(CO3)2(s) |
14.2 |
|
|
|
K+ + NpO2+ + CO32- ⇌ KNpO2(CO3)(s) |
13.6 |
|
|
|
3K+ + NpO2+ + 2CO32- + 0.5H2O ⇌ K3NpO2(CO3)2×0.5H2O(s) |
-4.8 |
|
|
Figure SOTERM-18. Predominant Species Diagram for Np as a Function of pH and Eh Based on the Np Speciation Data Reactions (SOTERM.60) to (SOTERM.70) (Richmann 2008)
The An(VI) FMT model has not been developed sufficiently for reliable use in predicting concentrations of this oxidation state in the WIPP brines under various solution conditions. Although uranyl carbonate can be successfully modeled, the hydrolysis behavior of U(VI) is quite complicated and no satisfactory predictive models applicable to WIPP-like conditions are yet available. Because the implementation of an MgO backfill limits the pmH and fCO2 to discrete values, empirical measurement of the solubility of U(VI) in WIPP and/or WIPP–like brines became practical. As documented in Hobart and Moore (1996) and used in prior PA calculations, the solubility of U(VI) at pH 10, in the absence of carbonate, was determined to be 8.8 ´ 10-6 m. This is augmented by additional data from U(VI) solubility studies in WIPP-relevant carbonate-free brines reported in Section SOTERM-3.3.2 (Lucchini et al. 2009). Here, the measured U(VI) solubility was 10-7 M to 10-6 M for GWB and ERDA-6 brine, respectively. The solubility of U(VI) currently used in WIPP PA was established through discussions with the EPA to be 1 mM (U.S. Environmental Protection Agency 2005) to account for the potential and expected effects of carbonate.
Details of the implementation of FMT and an early version of the CHEMDAT database are given in Novak (1995, Appendix D) and in the FMT User’s Manual (Babb and Novak 1995 and 1997). FMT calculates chemical equilibrium for user-specified total element amounts in aqueous or aqueous/mineral geochemical systems. The FMT calculations of actinide solubility in the WIPP system performed for WIPP PA included preequilibration with halite, anhydrite, brucite, and magnesite (Novak, Moore, and Bynum 1996; Novak and Moore 1996), which are the minerals present in large quantities in the repository. The effects of the MgO backfill are realized by equilibrating brine with brucite, magnesite, and hydromagnesite.
The Pitzer formalism is substantially different in approach from the classic Debye-Hückel (D-H) theory of the behavior of ionic solutions. The latter is a theoretical approach to describing the behavior of dilute solutions; more importantly, because many ionic solutes do not behave ideally even at very low concentrations, it provides a means to calculate the activity, ai, of a desired species. This is of great importance, as the Gibbs free energies of the various species in solution can be used to calculate solution equilibria if one knows the effective concentration of those species, i.e. their “activity” in solution. The activity of a given species i is tied to the molality of that species as ai = γimi. Since the molality of species i is known, the unknown that must be calculated to determine ai is, therefore, γi. The simplest form relating activity to molality from the D-H law is
where Aγ is the Debye-Hückel parameter, zi is the charge of the ith species and I is the overall solution ionic strength. The fundamental difficulty with the D-H formalism is that even with extensions (Davies equation, B-dot equation), the D-H law begins to deviate significantly from real solution behavior somewhere in the general region of I = 0.3 molal. As the WIPP brines (and many other highly concentrated ionic species of interest) are well above this level of ionic strength, many times with I > 5, another description is required to properly describe the activities of the ionic species.
In 1973, Pitzer proposed a set of semiempirical equations to describe ai. Pitzer (1973) wrote the Gibbs excess energy of a solution as a virial expansion, where a portion of the overall expansion can be tied down to a formalism similar to the D-H law and the majority of the remaining constants are empirically determined from measurements of the desired ions. The most general form of the equation is
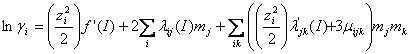 , (SOTERM.72)
, (SOTERM.72)
where f(I) is a Debye-Hückel function, f¢(I) is its derivative df/dI, the λij are second-order interaction coefficients, λ’ij(I) is the derivative dλij/dI, and the μijk are third-order interaction coefficients. The experimentally observable values β(0), β(1), β(2), α1, α2, Cφ, and so forth are used to calculate the λij and μijk values needed to calculate γi (for more detail, see Wolery and Daveler 1992).
This approach has proven highly effective and has successfully described the behavior of solutions at high ionic strength. The disadvantage of this technique is that binary and ternary coefficients for the expansion are normally needed to completely describe all the activities of the different species; in addition, if the number of species in solution grows, the number of calculations grows that much faster, i.e., on the order of the cube of the number of species. This problem would be even worse, except that many of the terms describing neutral species can be legitimately neglected in geochemical systems.
This parameter-determination problem is of particular interest in the description of actinide behavior in the WIPP, since the GWB and ERDA-6 brines of interest contain a wide variety of ions in and of themselves, in addition to the actinides introduced into the repository. As a result of this, it was necessary to constrain the total number of possible species in solution, aqueous, solid or gas, and in addition, to determine Pitzer parameters for many species by analogy to others rather than by experimental measurement. This is the basis of the parameter and species selection in the current database, FMT_050405.CHEMDAT, which contains the parameters (free energies, Pitzer parameters, etc.) for those species incorporated into the limited species set description. In practice, this has worked well to describe solution behavior in the WIPP within a limited set of pH values at 25 oC, but does not describe the WIPP system in all regions of interest.
The oxidation-state-specific actinide solubilities calculated with the FMT thermodynamic model are summarized in Table SOTERM-17 for the CRA-2004 PABC. For historical perspective, the calculated solubilities from prior PA analyses are tabulated in Table SOTERM-18. In the CRA-2004 PABC, the data are shown for two brines in the presence of organics, and as a function of equilibration with hydromagnesite or magnesite. The hydromagnesite case is recognized by the project as the most relevant to WIPP. It is important to note that, overall, the calculated solubilities have not changed much over time (generally within a factor of two) when organics are not considered.
Table SOTERM-17. Solubilities of the Oxidation-State Analogs (M) with MgO Backfill Calculated for the CRA-2004 PABC (Brush 2005)
|
Brine |
FMT Name |
Solubilities of the Actinide Oxidation States from the FMT Calculations for PABC |
|||
|
(III) |
(IV) |
(V) |
(VIa) |
||
|
GWB |
Run 7 (hydromagnesite with organics [hmag. w orgs.]) |
3.87 ´ 10-7 |
5.64 ´ 10-8 |
3.55 ´ 10-7 |
1 ´ 10-3 |
|
ERDA-6 |
Run 11 (hmag. w orgs) |
2.88 ´ 10-7 |
6.79 ´ 10-8 |
8.24 ´ 10-7 |
1 ´ 10-3 |
|
GWB |
Run 5 (mag. w orgs) |
3.87 ´ 10-7 |
4.57 ´ 10-8 |
6.59 ´ 10-6 |
1 ´ 10-3 |
|
ERDA-6 |
Run 9 (mag. w orgs) |
2.87 ´ 10-7 |
4.84 ´ 10-8 |
1.08 × 10-5 |
1 ´ 10-3 |
|
hmag. – hydromagnesite mag. – magnesite a Not calculated with the FMT model |
|||||
Table SOTERM-18. Historical Actinide Solubilities (M) Calculated (III, IV, and V) or Estimated (VI) for the CRA-2004 PA, the CCA PAVT and the CCA PA (U.S. Department of Energy 2004)
|
Actinide Oxidation State, and Brine |
CRA Solubilities, Microbial Vectors |
CRA Solubilities, Nonmicrobial Vectors |
PAVT Solubilities |
CCA Solubilities |
|
III, Salado brine |
3.07 ´ 10-7 |
3.07 ´ 10-7 |
1.2 ´ 10-7 |
5.82 ´ 10-7 |
|
III, Castile brine |
1.69 ´ 10-7 |
1.77 ´ 10-7 |
1.3 ´ 10-8 |
1.3 ´ 10-8 |
|
IV, Salado brine |
1.19 ´ 10-8 |
1.24 ´ 10-8 |
1.3 ´ 10-8 |
4.4 ´ 10-6 |
|
IV, Castile brine |
2.47 ´ 10-8 |
5.84 ´ 10-9 |
4.1 ´ 10-9 |
6.0 ´ 10-9 |
|
V, Salado brine |
1.02 ´ 10-6 |
9.72 ´ 10-7 |
2.4 ´ 10-7 |
2.3 ´ 10-6 |
|
V, Castile brine |
5.08 ´ 10-6 |
2.13 ´ 10-5 |
4.8 ´ 10-5 |
2.2 ´ 10-6 |
|
VI, Salado brine |
8.7 ´ 10-6 |
8.7 ´ 10-6 |
8.7 ´ 10-6 |
8.7 ´ 10-6 |
|
VI, Castile brine |
8.8 ´ 10-6 |
8.8 ´ 10-6 |
8.8 ´ 10-6 |
8.8 ´ 10-6 |
The calculated solubility of the III actinides was 2.87 ´ 10-7 M to 3.87 ´ 10-7 M in the CRA-2004 PABC (Brush and Xiong 2005b). These data are also fairly consistent with recently measured results for Nd(III) solubility in brine (Borkowski et al. 2008). A somewhat broader range was noted historically: 1.3 ´ 10-8 M to 5.82 ´ 10-7 M. The expected solubility of the IV actinides ranges between 4.57 ´ 10-8 M and 6.79 ´ 10-8 M. This is also somewhat consistent with prior calculations (Table SOTERM-18) and has increased slightly. Overall the solubility of the IV actinides is four to eight times lower than that predicted for the III actinides. The main reason for increases noted in CRA-2004 PABC was the presence of organics in the brines.
Uncertainties in the solubility data and uncertainty in the NONLIN least-squares refinement, for Pitzer parameter determination, result in uncertainty in the model predictions. This distribution was sampled and used in PA as discussed in Section SOTERM-5.0 (Xiong, Nowak, and Brush 2005).
Four organic ligands are included in FMT calculations of actinide solubilities. These are acetate (CH3CO2-), citrate [(CH2CO2)2C(OH)(CO2) 3-], EDTA [(CH2CO2)2N(CH2)2N(CH 2CO2)24-], and oxalate (C2O42-). The current projected inventory of these complexing agents, with their inventory-limited solubilities in the WIPP, were summarized in Table SOTERM-5. These ligands are included in the solubility calculations because (1) approximately 60 organic compounds were identified among the nonradioactive constituents of the TRU waste to be emplaced in the WIPP (Brush 1990; Drez 1991; U.S. Department of Energy 1996); (2) 10 of these 60 organic compounds could, if present in the WIPP, increase actinide solubilities because they are soluble in aqueous solutions such as WIPP brines, and because they form complexes with dissolved actinides (Choppin 1988); and (3) of these 10 water-soluble organic ligands that form complexes with actinides, 4 (acetate, citrate, EDTA, and oxalate) were identified in the WIPP inventory (See the CCA, Appendix SOTERM, p. 96).
The effects of all four ligands (acetate, citrate, EDTA, and oxalate), as well as the Mg2+ and Ca2+ species present in brine, were addressed in the calculations of actinide solubility for GWB and ERDA-6 brine in the PABC calculations (Brush 2005). The stability constants for the complexes formed by the listed ligands with Ca2+ were assigned the same values as the stability complexes formed by these ligands with Mg2+ (Giambalvo 2003). Because of insufficient data these calculations did not include competition from the other dissolved metals such as Fe, V, Cr, Ni, copper (Cu), and Pb, all of which could be present at significant concentrations due to dissolution of steels and other metallic constituents of TRU waste (see Table SOTERM-19 and U.S. Department of Energy 2006). The FMT calculations (Brush 2005) demonstrate that the solubility of the III and IV actinides was not significantly enhanced by complexation with acetate, citrate, EDTA, and oxalate at their maximum potential concentrations (Table SOTERM-19). EDTA does, however, exert a strong influence on the speciation of the III actinides, in that it essentially forms a 1:1 complex with the actinide. In this context, higher levels of EDTA in the repository, should they exist, could overwhelm carbonate complexation and hydrolysis to dominate the speciation of the III actinides.
In the FMT calculations, all four ligands (acetate, citrate, EDTA, and oxalate) were present simultaneously in Salado or Castile brine at the concentrations calculated by Brush and Xiong (2005a). The results of the FMT calculations for the CRA-2004 PABC demonstrate that acetate, citrate, EDTA, and oxalate will not form complexes with the III and IV actinides to a significant extent under expected WIPP conditions, and thus will not significantly affect the III and IV actinide solubilities (Brush and Xiong 2003c; Downes 2003a and 2003b).
The
importance and role of colloids in defining the concentration of
actinide in the WIPP was discussed in Section SOTERM-3.8, and more extensive
discussions of WIPP-related results were
Table SOTERM-19. Comparison of Actinide Solubility Calculations With and Without Organics
|
Property or Actinide Oxidation State |
FMT Run 7 (GWB, hmag, with orgs) |
FMT |
FMT Run 11 (ERDA-6, hmag, with orgs) |
FMT Run 12 (ERDA-6, hmag, without orgs) |
|
An(III), M |
3.87 × 10-7 |
2.26 × 10-7 |
2.88 × 10-7 |
8.67 × 10-8 |
|
An(IV), M |
5.64 × 10-8 |
5.66 × 10-8 |
6.79 × 10-8 |
7.20 × 10-8 |
|
An(V), M |
3.55 × 10-7 |
2.36 × 10-7 |
8.24 × 10-7 |
5.38 × 10-7 |
|
I, m |
7.66 |
7.54 |
6.80 |
6.72 |
|
log fCO2 |
-5.50 |
-5.50 |
-5.50 |
-5.50 |
|
ρ, kg/m3 |
1230 |
1230 |
1220 |
1220 |
|
pH |
8.69 |
8.69 |
8.94 |
9.02 |
|
RH, % |
73.2 |
73.3 |
74.8 |
74.8 |
presented in the CRA-2004, Appendix PA, Attachment SOTERM. Results of the colloidal actinide investigation were used in the CRA-2004 PABC, the CRA-2004 PA, the CCA PA, and the 1997 PAVT to define the PA approach to accounting for colloidal enhancement of actinide concentrations. The four types of colloids identified as relevant to the WIPP are listed and described in Table SOTERM-20.
Table SOTERM-20. Classification of Four Colloid Types Considered by WIPP PA
|
Mineral Fragment Colloids |
Hydrophobic, hard-sphere particles that are kinetically stabilized or destabilized by electrostatic forces and may consist of crystalline or amorphous solids. Mineral fragments may be made kinetically stable by coatings with steric stabilizers that prevent close contact. Mineral fragments may act as substrates for sorption of actinides, or they may consist of precipitated or coprecipitated actinide solids. |
|
Intrinsic Actinide Colloids |
Intrinsic actinide colloids (also known as true colloids, real colloids, Type I colloids, and Eigenkolloide) are macromolecules of actinides that, at least in some cases, may mature into a mineral-fragment type of colloidal particle. When immature, they are hydrophilic; when mature, they become hydrophobic. |
|
Humic Colloids |
Humic substances are hydrophilic, soft-sphere particles that are stabilized by solvation forces. They are often powerful substrates for uptake of metal cations and are relatively small (less than 100,000 atomic mass units). |
|
Microbial Colloids |
Microbes are relatively large colloidal particles stabilized by hydrophilic coatings on their surfaces, which behave as steric stabilizing compounds. They may act as substrates for extracellular actinide sorption or actively bioaccumulate actinides intracellularly. |
Three types of parameter values were determined: (1) constant concentration values, (2) concentration values proportional to the dissolved actinide concentration, and (3) maximum concentration values. The parameter types are summarized below and were initially described in parameter record packages (Papenguth and Behl 1996a; Papenguth 1996a, 1996b, and 1996c) and resummarized for the CRA-2004 PABC (Garner and Leigh 2005). For intrinsic actinide colloids and mineral-fragment colloids, associated actinide concentrations were described as constant values. Table SOTERM-21 summarizes the material and parameter names and descriptions.
Table SOTERM-21. Material and Property Names for Colloidal Parameters
|
Material |
Property |
Brief Description of Parameter |
|
Th, U, Np, Pu, Am |
CONCMIN |
Concentration of actinide associated with mobile mineral fragment colloids |
|
Th, U, Np, Pu, Am |
CONCINT |
Concentration of actinide associated with mobile intrinsic actinide colloids |
|
Th, U, Np, Pu, Am |
PROPMIC |
Proportionality constant for concentration of actinides associated with mobile microbes |
|
PHUMOX3a |
PHUMCIM |
Proportionality constant for concentration of actinides associated with mobile humic colloids; in Castile brine; actinide solubilities are inorganic only (complexes with man-made organic ligands are not important); solubilities were calculated assuming equilibrium with Mg-bearing minerals (brucite and magnesite) |
|
PHUMOX3a PHUMOX6 |
PHUMSIM |
Proportionality constant for concentration of actinides associated with mobile humic colloids; in Salado brine; actinide solubilities are inorganic only (complexes with man-made organic ligands are not important); solubilities were calculated assuming equilibrium with Mg-bearing minerals (brucite and magnesite) |
|
Th, U, Np, Pu, Am |
CAPMIC |
Maximum (cap) concentration of actinide associated with mobile microbes |
|
Th, U, Np, Pu, Am |
CAPHUM |
Maximum (cap) concentration of actinide associated with mobile humic colloids |
|
a Proportionality constant for actinide concentrations associated with mobile humic substances for PHUMOX3, for actinide elements with oxidation state III (that is, Pu(III) and Am(III)); PHUMOX4, oxidation state IV (Th(IV), U(IV), Np(IV), and Pu(IV)); PHUMOX5, oxidation state V (Np(V)); and PHUMOX6, oxidation state VI (U(VI)). |
||
Experiments conducted to quantify actinide concentrations associated with humic substances and microbes provided the basis for a more sophisticated representation, in which colloidal actinide concentrations were related to the dissolved actinide concentration by proportionality constants. For microbes, the proportionality relationship was made by element. For humic actinides, however, the relationship was made by oxidation state, rather than by element. For microbes and humic substances, the experiments also provided a basis to define upper limits of the actinide concentration that could be associated with each of those colloid types. For both humic and microbial actinides, the upper limit parameter was defined by element, rather than oxidation state, and is in units of molality. The use of the two upper limit parameters is slightly different, and is described in the sections below discussing humic substances and microbes.
The colloid concentration factors used in the
CRA-2004 PABC are summarized in Table SOTERM-22. The general approach used to account
for colloidal enhancement of actinide solubilities is described
in detail by Garner and Leigh (2005). There were essentially no changes in the
approach used from the CRA-2004 PA. The maximum
concentrations of actinides predicted for the four types of WIPP
colloids are tabulated in Table SOTERM-23. These data
Table SOTERM-22. Colloid Concentration Factors (The CRA-2004, Appendix PA, Attachment SOTERM)
|
Actinide |
CONCMIN |
CONCINT |
PROPMIC |
CAPMIC |
Proportion Sorbed on Humicsb |
CAPHUM |
|
|
PHUMSIM |
PHUMCIM |
||||||
|
Th(IV) |
2.6 ´ 10-8 |
0.0 |
3.1 |
0.0019 |
6.3 |
6.3 |
1.1 ´ 10-5 |
|
U(IV) |
2.6 ´ 10-8 |
0.0 |
0.0021 |
0.0021 |
6.3 |
6.3 |
1.1 ´ 10-5 |
|
U(VI) |
2.6 ´ 10-8 |
0.0 |
0.0021 |
0.0021 |
0.12 |
0.51 |
1.1 ´ 10-5 |
|
Np(IV) |
2.6 ´ 10-8 |
0.0 |
12.0 |
0.0027 |
6.3 |
6.3 |
1.1 ´ 10-5 |
|
Np(V) |
2.6 ´ 10-8 |
0.0 |
12.0 |
0.0027 |
9.1 ´ 10-4 |
7.4 ´ 10-3 |
1.1 ´ 10-5 |
|
Pu(III) |
2.6 ´ 10-8 |
1.0 ´ 10-9 |
0.3 |
6.8 ´ 10-5 |
0.19 |
1.37 e |
1.1 ´ 10-5 |
|
Pu(IV) |
2.6 ´ 10-8 |
1.0 ´ 10-9 |
0.3 |
6.8 ´ 10-5 |
6.3 |
6.3 |
1.1 ´ 10-5 |
|
Am(III) |
2.6 ´ 10-8 |
0.0 |
3.6 |
1.0 |
0.19 |
1.37 e |
1.1 ´ 10-5 |
|
a In units of moles colloidal actinide per liter b In units of moles colloidal actinide per mole dissolved actinide c For the CRA-2004 PABC, all vectors were microbial d In units of moles total mobile actinide per liter e A cumulative distribution from 0.065 to 1.60 with a median value of 1.37 was used NOTE: The colloidal source term is added to the dissolved source term to arrive at a total source term. Mineral fragments were provided with distributions, but the maximum was used as described in the CRA-2004, Appendix PA, Attachment SOTERM. Humic proportionality constants for the III, IV, and V states were provided with distributions, but only the Castile Am(III) and Pu(III) were sampled. |
|||||||
Table SOTERM-23. Actinide Concentration or Maximum Concentration Due to Colloidal Enhanced Solution Concentrations (Garner and Leigh 2005)
|
Actinide |
CAPHUM |
CAPMIC Microbial Colloids |
CONCMIN |
CONCINT |
PROPMIC |
|
1.1 ´ 10-5 M |
1.0 M |
2.6 ´ 10-8 M |
|||
|
1.1 ´ 10-5 M |
0.0027 M |
2.6 ´ 10-8 M |
2.7 ´ 10-3 |
||
|
1.1 ´ 10-5 M |
6.8 ´ 10-5 M |
2.6 ´ 10-8 M |
1.00 ´ 10-9 M |
6.8 ´ 10-5 |
|
|
1.1 ´ 10-5 M |
0.0019 M |
2.6 ´ 10-8 M |
1.9 ´ 10-3 |
||
|
1.1 ´ 10-5 M |
0.0021 M |
2.6 ´ 10-8 M |
2.1 ´ 10-3 |
||
|
a In units of moles colloidal actinide per mole dissolved actinide |
|||||
show that microbial colloids are likely to have the most significant effect on actinide concentrations, with a smaller but significant contribution from the humic colloidal fraction. Section SOTERM-5.0 provides more details on the PA implementation of these data.
The WIPP ASTP provided the parameters to construct the maximum dissolved and suspended colloidal actinide concentrations for use in modeling the mobilization and transport of actinides in the disposal system. In the WIPP PA, mobilization of radionuclides is represented by the PANEL code and transport of radionuclides within the repository and the Salado is represented by the Nuclide Transport System (NUTS) code (Appendix PA-2009, Section PA-4.4 and Section PA-4.3, respectively). A description of the simplifications, manipulations, and approach used in the PA to perform this modeling is discussed in this section.
The DOE has concentrated on those processes most likely to have a significant impact on system performance. Therefore, several simplifications were used in the modeling of radionuclide mobilization and transport in the CCA PA, the CCA PAVT, the CRA-2004 PA, the CRA-2004 PABC, and the CRA-2009 PA calculations. These include
· Using constant solubility parameters and constant colloidal parameters throughout the repository and regulatory period for a given realization
· Modeling only the isotopes most important to compliance
· Using the compositions of Castile and Salado brines (the end-member brines) to bracket the behavior of mixtures of these brines within the repository
· Sampling only the uncertain parameters with the most significant effect on repository performance
· Combining dissolved and colloidal species for transport within the disposal system, as modeled by NUTS and PANEL
Selection of isotopes for modeling mobilization and transport in the disposal system with NUTS and PANEL is described in Appendix PA-2009, Section PA-4.3.3. Runs of PANEL, the PA code that computes total mobilized radionuclide concentrations, include 29 radionuclides in the decay calculations (Garner and Leigh 2005, Table 7 and Table 12). Runs of NUTS, the PA code that computes radionuclide transport within the Salado, are based on five radionuclides: (230Th, 234U, 238Pu, 239Pu, and 241Am) that represent groupings of radionuclides with similar decay and transport properties (Appendix PA-2009, Section PA-4.3.3). The number of radionuclides for transport calculations in NUTS has been reduced because calculations for the full WIPP inventory and decay chains would be very time consuming and because accurate results can be achieved with this limited set of radionuclides.
Transport calculations in the Culebra use a reduced set of four radionuclides: (230Th, 234U, 239Pu, and 241Am) for computational efficiency (Garner 1996). 238Pu has been omitted from transport in the Culebra because its short half-life (87.7 years) means that little 238Pu will enter the Culebra via brine flows up a borehole.
The general scenarios described in Appendix PA-2009, Section PA-2.3.2 and Section PA-3.10 and considered in the source term calculations may be categorized into three groups: (1) undisturbed performance (BRAGFLO S1 scenario); (2) intrusion through the repository and into the Castile, intersecting a pressurized brine reservoir (BRAGFLO S2, S3, and S6 scenarios); and (3) intrusion through the repository, but not into a pressurized brine reservoir (BRAGFLO S4 and S5 scenarios). The specific scenarios and the associated type of borehole intrusion considered by the WIPP PA are listed in Table SOTERM-24.
Table SOTERM-24. WIPP PA Modeling Scenarios for the CRA-2004 PABC (Garner and Leigh 2005; Leigh et al. 2005)
|
BRAGFLO |
Description |
Brine Used in PA |
|
S1 |
E0 (Undisturbed Repository) |
Salado (GWB) |
|
S2 |
E1 intrusion at 350 years penetrates the repository and a brine pocket |
Castile (ERDA-6) |
|
S3 |
E1 intrusion at 1000 years penetrates the repository and a brine pocket |
Castile (ERDA-6) |
|
S4 |
E2 intrusion at 350 years penetrates the repository (only) |
Salado (GWB) |
|
S5 |
E2 intrusion at 1000 years penetrates the repository (only) |
Salado (GWB) |
|
S6 |
E2 intrusion at 1000 years penetrates the repository (only); E1 intrusion at 2000 years penetrates the repository and a brine pocket |
Castile (ERDA-6) |
Brine may enter the repository from three sources, depending on the nature of the borehole intrusion. Under all scenarios, brine may flow from the surrounding Salado through the DRZ and into the repository in response to the difference between the hydraulic head in the repository and in the surrounding formation. For the BRAGFLO S2 through S6 scenarios, in which a borehole is drilled into the repository, brine may flow down the borehole from the Rustler and/or the Dewey Lake. For the BRAGFLO S2, S3, and S6 scenarios, in which a pressurized Castile brine reservoir is intercepted, brine from the Castile may flow up the borehole into the repository.
As mentioned in Section SOTERM-2.3.1, the brines in the Salado and Castile have different compositions and the actinides solubilities are somewhat different in each of these end-member compositions.
The composition of the more dilute groundwaters from the Rustler and Dewey Lake are expected to change rapidly upon entering the repository as a result of fast dissolution of host Salado minerals from the walls and floor of the repository. These minerals comprise about 90-95% halite and about 1-2% each of polyhalite, gypsum, anhydrite, and magnesite (Brush 1990). Calculations titrating Salado rock into dilute brines using EQ3/6 (Wolery 1992; Wolery and Daveler 1992) show that gypsum, anhydrite, and magnesite saturate before halite. When halite saturates, the brine composition is very similar to that of Castile brine. One hundred times as much polyhalite must be added to the system before the resulting brine has a composition similar to Salado brines. These calculations indicate that if dilute brines dissolve away only the surfaces of the repository, they will obtain Castile-like compositions, but if they circulate through the Salado after saturating with halite, they may obtain compositions similar to Salado brine. Similarly, if Castile brine circulates through enough host rock, it may also approach Salado brine composition. In either case, the actual brine within the repository may be described as a mixture of the two concentrated-brine end members: Salado and Castile. This mixture, however, is very hard to quantify, because it is both temporally and spatially variable. Only in the undisturbed scenario is the mixture well defined as 100% Salado brine over the 10,000-year regulatory period. In this context, the Salado (GWB) and Castile (ERDA-6) brines bracket the range of expected brine compositions.
For a panel intersected by a borehole, the BRAGFLO calculations show that in the 10% of the repository represented by the BRAGFLO panel computational cells, the ratio of brine inflow that enters through the borehole versus through inflow from the host rock varies in time and depends on the sampled parameter values and scenario considered. This ratio was the only measure of brine mixing available to the source term runs in the CCA PA, the CCA PAVT, the CRA-2004 PA, and the CRA-2004-PABC calculations. As an estimate, this ratio (1) does not account for compositional changes that occur when H2O is consumed by corrosion reactions; (2) does not resolve the details of flow, diffusion, and brine interaction with internal pillars and the DRZ; and (3) is an average over one-tenth of the repository. It is expected that the fraction of Salado brine will be quite high in areas of the repository distant from the borehole and much lower near the borehole. Because radionuclide travel up the borehole can lead to significant release, the solubility of radionuclides near the borehole is important. Given these uncertainties, the DOE decided to use the Castile end-member composition to calculate radionuclide solubilities for scenarios where a borehole penetrates a brine reservoir, and to use the Salado end-member composition for scenarios where it does not (see Table SOTERM-24).
The parameters to be sampled for the PA were selected based on the expected significance of their effect on repository performance. The following four parameters are sampled independently (Garner and Leigh 2005, Table 3 and Table 8):
· The solubility uncertainty for oxidation state III (see discussion below and Figure SOTERM-19).
· The solubility uncertainty for oxidation state IV (see discussion below and Figure SOTERM-20).
Figure SOTERM-19. Frequency Distribution of the Deviation of Experimental log Solubility from Model-Predicted Value for all An(III) Comparisons. A Total of 243 Measured and Predicted Solubilities were Compared (Xiong 2005).
· The oxidation state for Pu, Np, and U. The sampled value is a flag that is “low” 50% of the time and “high” 50% of the time. If the flag is set to “high,” Pu is assumed to be in the IV oxidation state, Np is assumed to be in the V oxidation state, and U is assumed to be in the VI oxidation state. If the flag is set to “low,” Pu is assumed to be in the III oxidation state and Np and U are assumed to be in the IV oxidation state.
· The humic-acid proportionality constant for the III oxidation state in Castile brine (see Table SOTERM-22 and Figure SOTERM-21).
As discussed by Garner and Leigh (2005, Section 2.3), the solubility uncertainty for oxidation state V is zero. The solubility uncertainty for oxidation state VI is zero because the EPA specified a fixed, maximum solubility of 1 × 10-3 mol/L for U(VI).
Actinide solubilities for a single realization in the PA depend on (1) the oxidation state; (2) the brine for that realization (see Table SOTERM-24); and (3) the solution concentration uncertainty, as shown in Equation (SOTERM.73).
Figure SOTERM-20. Frequency Distribution of the Deviation of Experimental log Solubility from Model-Predicted Value for all An(IV) Comparisons. A Total of 45 Measured and Predicted Solubilities were Compared (Xiong 2005).
Ci,b = (Si,b) ´ (10SUi) (SOTERM.73)
Ci,b, used for every element in oxidation state i, is the concentration of oxidation state i and brine b. Si,b is the solubility calculated for oxidation state i in brine b with FMT (see Table SOTERM-17). SUi is the solubility uncertainty sampled from a distribution unique to each oxidation state. Figure SOTERM-19 shows the distribution of SU values for oxidation state III. Figure SOTERM-20 shows the distribution of SU values for oxidation state IV. These distributions are calculated and documented in Xiong (2005).
Figure SOTERM-21 shows the cumulative distribution function for the humic-acid proportionality constant.
Figure SOTERM-21. Cumulative Distribution Function for the Humic-Acid Proportionality Constant
Dissolved and colloidal species may transport differently because of different diffusion rates, sorption onto stationary materials, and size-exclusion effects (filtration and hydrodynamic chromatography). With maximum molecular diffusion coefficients of about 4 ´ 10-10 m2/s, actinides are estimated to diffuse about 10 m in 10,000 years, a negligible distance. Sorption and filtration have beneficial but unquantified effects on performance. Hydrodynamic chromatography may increase colloidal transport over dissolved transport by, at most, a factor of two for theoretically perfect colloidal-transport conditions. In the WIPP, the expected increase is much lower. Given the small or beneficial nature of these effects, they were not included in the CCA PA, the CCA PAVT, the CRA-2004 PA, or the CRA-2004 PABC calculations of radionuclide transport in the repository.
Because there was no modeled mechanism in PA to differentiate dissolved from colloidal species, the DOE combined them for transport within the Salado. To model transport within the Culebra, however, this simplification was replaced by separating the mobilized actinides delivered to the Culebra by Salado transport codes into five components (dissolved, humic, microbial, mineral-fragment, and intrinsic colloids) to account for differences in their transport behavior. This is important because transport within the repository occurs through, at most, hundreds of meters of poorly defined waste undergoing decomposition, whereas transport through the Culebra occurs over kilometers in a relatively homogeneous (compared to waste) fractured dolomite.
The parameters required to construct the source term were as follows:
1. Solubilities for four oxidation states in Salado and Castile brines, the two brine end members.
2. Uncertainty distributions to be applied to the median solubilities for oxidation states III and IV.
3. A scheme for assigning sampled oxidation states (“low” or “high”).
4. Colloidal concentrations or proportionality constants for each actinide (Th, U, Np, Pu, and Am) and an associated oxidation state for each of four colloid types.
5. Caps on the actinide concentrations that may be applied to two types of colloids (microbial and humic).
6. Cm is assigned the source term calculated for Am.
Cm and Np are not explicitly transported in NUTS (see Section SOTERM-5.1.1) although they are implicitly lumped with other modeled isotopes. They are, however, included in the PANEL calculations for use with the DBR calculations in PA.
These parameters are combined into a single maximum concentration for each modeled actinide in the PA calculations. The term “total mobilized concentration” is used for the combined concentrations of dissolved and colloidal species. The combined concentrations are not necessarily the actual concentrations, because the concentration may be lower as a result of inventory limits. Both NUTS and PANEL assume that the actinide concentrations specified by the total mobilized concentrations are attained instantaneously as long as sufficient inventory is available. When the inventory is insufficient, the actual mobilized concentration will be lower and is said to be inventory limited. The calculation of the total mobilized concentration is performed by PANEL for each of 100 sampled vectors in a replicate. A similar methodology to generate the combined maximum concentrations was also used for the CCA PA, the CCA PAVT, the CRA-2004 PA, and the CRA-2004 PABC.
All of the source term parameters and their associated distributions are entered into the PA parameter database. For each sampled parameter, the Latin Hypercube Sampling code uses the distribution from the PA parameter database to create 100 sampled values. These values are combined with the parameters that have constant values and stored in computational databases for each of the 100 vectors (i.e., 100 realizations), which constitute one replicate. For each realization, PANEL uses both the constant and sampled values for all of the source term parameters, and constructs the source term for NUTS and PANEL, as shown below. This process is repeated for scenarios using the Salado end-member total mobilized concentration and for scenarios using the Castile end-member total mobilized concentration.
Dissolved = Base Solubility × 10 Sampled from Solubility Distribution (SOTERM.74)
IF (Dissolved × Proportionality
Constant < Humic Cap),
THEN Humic = Dissolved × Proportionality Constant,
(SOTERM.75)
ELSE Humic = Humic
Cap
IF (Total Mobile < Microbial Cap),
THEN Microbial = Dissolved × Proportionality
Constant, (SOTERM.76)
ELSE Microbial = Microbial
Cap
Mineral = Database Concentration (a constant value) (SOTERM.77)
Intrinsic = Database Concentration (a constant value) (SOTERM.78)
Total Mobile = Dissolved + Humic + Microbial + Mineral + Intrinsic (SOTERM.79)
For actinides with more than one oxidation state, the oxidation state is specified by the oxidation-state parameter
IF (OXSTAT £ 0.5); THEN Lower
Oxidation State,
ELSE Higher Oxidation
State
(SOTERM.80)
where OXSTAT is the oxidation-state parameter sampled from a uniform distribution between 0 and 1.
Similar solubility calculations are performed for Am, U, Th, and Np. The total mobilized concentration and mobile fractions for Cm are set equal to the values for Am. In addition, the PA groups radioisotopes with similar decay and transport properties for the NUTS and SECOTP2D (component radionuclide transport in fractures or granular acquifers) transport calculations, as explained in Section SOTERM-5.1.1. For example, the U solubility is decreased to account for the shared solubility with the low-activity 238U, which is not explicitly modeled, enabling NUTS to properly represent the effect of the U isotopes on compliance using the single lumped isotope 234U (the CRA-2004, Appendix PA, Section PA-4.3.3).
PANEL also calculates the fraction of each actinide mobilized by the five different mechanisms, as follows:
Fraction dissolved = Dissolved/Total Mobilized Concentration (Conc.) (SOTERM.81)
Fraction on humics = Humic/Total Mobilized Conc. (SOTERM.82)
Fraction in/on microbes = Microbe/Total Mobilized Conc. (SOTERM.83)
Fraction on mineral fragments = Mineral/Total Mobilized Conc. (SOTERM. 84)
Fraction as intrinsic colloid = Intrinsic/Total Mobilized Conc. (SOTERM.85)
For example, for one realization in Salado brine, the sampled value for OXSTAT was 0.9, so Pu would be present in the IV state. The sampled value of the solubility uncertainty distribution was 1.8 for the IV state, which has a median brine solubility of 5.64 ´ 10-8 M. The humic proportionality constant for the IV oxidation state in Salado brine is 6.3, the microbial proportionality constant for Pu is 0.3, the humic cap is 1.1 ´ 10-5 M, the microbe cap for Pu is 6.8 ´ 10-5 M, the concentration of the actinide on mineral fragments is 2.6 ´ 10-8 M, and the Pu intrinsic-colloid concentration is 1 ´ 10-9 M.
For this realization, the maximum dissolved concentration of Pu(IV) used by the PA would be
CPu = (5.64 ´ 10-8) ´ (101.8) = 3.6 ´ 10-6 M. (SOTERM.86)
(The calculations for this example have been rounded to two significant figures, although the PA would not round the intermediate or final values.) CPu is the maximum dissolved concentration of all combined isotopes of Pu.
The maximum humic-complexed Pu would be
(3.6 ´ 10-6 M)(6.3 mol adsorbed per mol) = 2.3 ´ 10-5 M. (SOTERM.87)
This value, however, exceeds the cap for humic-mobilized Pu, 1.1 ´ 10-5 M. Therefore, in this case, the cap would be used for the maximum humic-mobilized actinide concentration. Note that the humic-mobilized concentration of Pu exceeds the maximum dissolved concentration of Pu, which is usually the case.
The maximum microbial-mobilized Pu would be
(3.6 ´ 10-6 M)(0.3 mol bioaccumulated per mol) = 1.1 ´ 10-6 M. (SOTERM.88)
This value is less than the cap, 6.8 ´ 10-5 M, so the cap does not affect microbial-mobilized Pu for this realization.
The total mobilized concentration of Pu(IV) for this realization would then be the sum of the dissolved and colloidal contributions (see Equation [SOTERM.79]):
Total Mobile = Dissolved + Humic + Microbial + Mineral +
Intrinsic, (SOTERM.89)
= 3.6
´ 10-6 +
1.1 ´
10-5 + 1.1 ´ 10-6 + 2.6
´
10-8 + 1.0 ´ 10-9,
= 1.6
´ 10-5
M.
The output of the PANEL calculations is a
computational database containing the source term and effective
inventories. NUTS and PANEL both assume instantaneous
dissolution and colloidal mobilization up to the solubility
limits when sufficient inventory is present, as discussed in Appendix PA-2009,
Section PA-4.3. Table SOTERM-25 shows the dissolved and
Table SOTERM-25. Concentrations (M) of Dissolved, Colloidal, and Total Mobile Actinides Obtained Using Median Parameter Values for the CCA PAVT and CRA-2004 PABCa
|
Actinide Oxidation State and Brine |
PAVT |
CRA-2004 PABC |
|
Pu(III), dissolved, Salado brine |
9.75 × 10-8 |
3.61 × 10-7 |
|
Pu(III), colloidal, Salado brine |
7.48 × 10-8 |
2.04 × 10-7 |
|
Pu(III), total mobile, Salado brine |
1.72 × 10-7 |
5.64 × 10-7 |
|
Pu(III), dissolved, Castile brine |
1.06 × 10-8 |
2.68 × 10-7 |
|
Pu(III), colloidal, Castile brine |
4.46 × 10-8 |
4.75 × 10-7 |
|
Pu(III), total mobile, Castile brine |
5.52 × 10-8 |
7.44 × 10-7 |
|
Am(III), dissolved, Salado brine |
9.75 × 10-8 |
3.61 × 10-7 |
|
Am(III), colloidal, Salado brine |
3.96 × 10-7 |
1.39 × 10-6 |
|
Am(III), total mobile, Salado brine |
4.93 × 10-7 |
1.75 × 10-6 |
|
Am(III), dissolved, Castile brine |
1.06 × 10-8 |
2.68 × 10-7 |
|
Am(III), colloidal, Castile brine |
7.78 × 10-8 |
1.34 × 10-6 |
|
Am(III), total mobile, Castile brine |
8.83 × 10-8 |
1.61 × 10-6 |
|
Th(IV), dissolved, Salado brine |
1.06 × 10-8 |
6.70 × 10-8 |
|
Th(IV), colloidal, Salado brine |
1.25 × 10-7 |
6.56 × 10-7 |
|
Th(IV), total mobile, Salado brine |
1.36 × 10-7 |
7.23 × 10-7 |
|
Th(IV), dissolved, Castile brine |
3.33 × 10-8 |
8.07 × 10-8 |
|
Th(IV), colloidal, Castile brine |
3.39 × 10-7 |
7.85 × 10-7 |
|
Th(IV), total mobile, Castile brine |
3.73 × 10-7 |
8.65 × 10-7 |
|
U(IV), dissolved, Salado brine |
1.06 × 10-8 |
6.70 × 10-8 |
|
U(IV), colloidal, Salado brine |
9.26 × 10-8 |
4.48 × 10-7 |
|
U(IV), total mobile, Salado brine |
1.03 × 10-7 |
5.15 × 10-7 |
|
U(IV), dissolved, Castile brine |
3.33 × 10-8 |
8.07 × 10-8 |
|
U(IV), colloidal, Castile brine |
2.36 × 10-7 |
5.35 × 10-7 |
|
U(IV), total mobile, Castile brine |
2.69 × 10-7 |
6.15 × 10-7 |
|
Pu(IV), dissolved, Salado brine |
1.06 × 10-8 |
6.70 × 10-8 |
|
Pu(IV), colloidal, Salado brine |
9.67 × 10-8 |
4.69 × 10-7 |
|
Pu(IV), total mobile, Salado brine |
1.07 × 10-7 |
5.36 × 10-7 |
|
Pu(IV), dissolved, Castile brine |
3.33 × 10-8 |
8.07 × 10-8 |
|
Pu(IV), colloidal, Castile brine |
2.47 × 10-7 |
5.60 × 10-7 |
|
Pu(IV), total mobile, Castile brine |
2.80 × 10-7 |
6.40 × 10-7 |
|
U(VI), dissolved, Salado brine |
7.07 × 10-6 |
1.00 × 10-3 |
|
U(VI), colloidal, Salado brine |
8.89 × 10-7 |
1.31 × 10-5 |
|
a Values are calculated using data retrieved from the WIPP PA Database http://yardbirds.sandia.gov/pview/ and equations SOTERM.75 through SOTERM.79. |
||
Table SOTERM-25. Concentrations (M) of Dissolved, Colloidal, and Total Mobile Actinides Obtained Using Median Parameter Values for the CCA PAVT and CRA-2004 PABCa (Continued)
|
Actinide Oxidation State and Brine |
PAVT |
CRA-2004 PABC |
|
U(VI), total mobile, Salado brine |
7.96 × 10-6 |
1.01 × 10-3 |
|
U(VI), dissolved, Castile brine |
7.15 × 10-6 |
1.00 × 10-3 |
|
U(VI), colloidal, Castile brine |
3.69 × 10-6 |
1.31 × 10-5 |
|
U(VI), total mobile, Castile brine |
1.08 × 10-5 |
1.01 × 10-3 |
|
a Values are calculated using data retrieved from the WIPP PA Database http://yardbirds.sandia.gov/pview/ and equations SOTERM.75 through SOTERM.79. |
||
colloidal components of the source term and the total mobile actinide concentrations obtained when median parameter values are used. The values from CRA-2004 PABC have been used as the source term in the PA for CRA-2009.
Abdelouas, A., W. Lutze, W. Gong, E.H. Nuttall, B.A. Strietelmeier, and B.J. Travis. 2000. “Biological Reduction of Uranium in Groundwater and Subsurface Soil.” The Science of the Total Environment, vol. 250: 21...\..\references\Others\Abdelouas_et_al_2000_Bio_Reduction_of_U_in_Groundwater.pdf
Allard, B. 1982. “Solubilities of Actinides in Neutral or Basic Solutions.” Actinides in Perspective (pp. 553–80). N. Edelstein, ed. New York: Pergamon...\..\references\Others\Allard_1982_Actinides_in_Perspective.pdf
Altmaier, M., V. Metz, V. Neck, R. Muller, and T. Fanghänel. 2003. “Solid-Liquid Equilibria of Mg(OH)2(cr) and Mg2(OH)3Cl4H2O(cr) in the System Mg-Na-H-OH-Cl-H2O at 25ºC.” Geochimica et Cosmochimica Acta, vol. 67: 3595–3601...\..\references\Others\Altmaier_Metz_Neck_Muller_Fanghanel_2003_Solid_Liquid_Equilibria.pdf
Altmaier, M., V. Neck, and Th. Fanghänel. 2004. “Solubility and Colloid formation of Th(IV) in concentrated NaCl and MgCl2 Solutions.” Radiochimica Acta, vol. 92: 537–43...\..\references\Others\Altmaier_Neck_Fanghanel_2004_Solubility_and_Colloid_Formation.pdf
Altmaier, M., V. Neck, R. Müller, and Th. Fanghänel. 2005. “Solubility of ThO2 xH2O(am) in Carbonate Solution and the Formation of Ternary Th(IV) Hydroxide-carbonate Complexes.” Radiochimica Acta, vol. 93: 83–92...\..\references\Others\Altmaier_Neck_Muller_Fanghanel_2005_Solubility_of_ThO2_xH2O.pdf
Altmaier, M., V. Neck, M.A. Denecke, R. Yin, and Th. Fanghänel. 2006. “Solubility of ThO2 ×xH2O(am) and the Formation of Ternary Th(IV) Hydroxide-Carbonate Complexes in NaHCO3-Na2CO3 Solutions Containing 0–4M NaCl.” Radiochimica Acta, vol. 94: 495–500...\..\references\Others\Altmaier_Neck_Denecke_Yin_Fanghanel_2006_Solubility_of_ThO2_xH2O.pdf
Altmaier, M., V. Neck, and Th. Fanghänel. 2007. Solubility of Zr(IV), Th(IV) and Pu(IV) Hydrous Oxides in CaCl2 Solutions and the Formation of Ternary Ca-M(IV)-OH Complexes. Manuscript presented at Migration 2007 Conference. 26-31 August 2007. Munich: Germany...\..\references\Others\Altmaier_Neck_Fanghanel_2007_Solubility_of_ZrIV_ThIV.pdf
Asbury, S.M., S.P. Lamont, and S.B. Clark. 2001. “Plutonium Partitioning to Colloidal and Particulate Matter in an Acidic, Sandy Sediment: Implications for Remediation Alternatives and Plutonium Migration.” Environmental Science and Technology, vol. 35: 2295–2300...\..\references\Others\Asbury_Lamont_Clark_2001_Pu_Partitioning_to_Colloidal.pdf
Babb, S.C., and C.F. Novak. 1995. User’s Manual for FMT, Version 2.0. ERMS 228119. Albuquerque: Sandia National Laboratories, WIPP Performance Assessment...\..\references\Others\Babb_Novak_1995_Users_Manual_for_FMT_Version_20.pdf
Babb, S.C., and C.F. Novak. 1997. User’s Manual for FMT, Version 2.3: A Computer Code Employing the Pitzer Activity Coefficient Formalism for Calculating Thermodynamic Equilibrium in Geochemical Systems to High-Electrolyte Concentrations. ERMS 243037. Albuquerque: Sandia National Laboratories, WIPP Performance Assessment...\..\references\Others\Babb_Novak_1997_Users_Manual_for_FMT_Version_23A.pdf
Banaszak, J.E., D.T. Reed, and B.E. Rittmann. 1998. “Speciation-Dependent Toxicity of Neptunium(V) Towards Chelatobacter heintzii.” Environmental Science and Technology, vol. 32: 1085–91...\..\references\Others\Banaszak_Reed_Rittmann_1998_Speciation_Dependent_toxicity_of_NpV.pdf
Banaszak, J.E., B.E. Rittmann, and D.T. Reed. 1998. Subsurface Interactions of Actinide Species and Microorganisms: Implications for the Bioremediation of Actinide-Organic Mixtures. ANL-98/26. Argonne, IL: Argonne National Laboratory...\..\references\Others\Banaszak_Rittmann_Reed_1998_Subsurface_Interactions_of_An_Species.pdf
Banaszak, J.E., S.M. Webb, B.E. Rittmann, J.-F. Gaillard, and D.T. Reed. 1999. “Fate of Neptunium in Anaerobic, Methanogenic Microcosm.” Scientific Basis for Nuclear Waste Management XXII, vol. 556: 1141–49...\..\references\Others\Banaszak_et_al_1999_Fate_of_Np_in_Anaerobic.pdf
Barton, L.L., K. Choudhury, B.M. Thomson, K. Steenhoudt, and A.R. Groffman. 1996. “Bacterial Reduction of Soluble Uranium: The First Step of In Situ Immobilization of Uranium.” Radioactive Waste Management and Environmental Restoration, vol. 20: 141–51...\..\references\Others\Barton_et_al_1996_Bacterial_Reduction_of_Soluble_Uranium.pdf
Behrends, T., and P. Van Cappellen. 2005. Competition Between Enzymatic and Abiotic Reduction of Uranium(VI) under Iron Reducing Conditions. Chemical Geology, vol. 220: 315–27...\..\references\Others\Behrends_VanCappellen_2005_Comp_Between_Enzymatic_and_Abiotic_UVI.pdf
Bender, J., M.C. Duff, P. Phillips, and M. Hill. 2000. “Bioremediation and Bioreduction of Dissolved U(VI) by Microbial Mat Consortium Supported on Silica Gel Particles.” Environmental Science and Technology, vol. 34: 3235–41...\..\references\Others\Bender_et_al_2000_Bioremediation_and_Bioreduction_of_Dissolved_UVI.pdf
Borkowski, M., J.F. Lucchini, M.K. Richmann, and D.T. Reed. 2008. Actinide (III) Solubility in WIPP Brine: Data Summary and Recommendations. LCO-ACP-08, LANL\ACRSP Report. Los Alamos, NM: Los Alamos National Laboratory...\..\references\Others\LCO_ACP_08.pdf
Brendebach, B., M. Altmaier, J. Rothe, V. Neck, and M.A. Denecke. 2007. “EXAFS Study of Aqueous Zr(IV) and Th(IV) Complexes in Alkaline CaCl2 Solutions: Ca3[Zr(OH)6]4+ and Ca4[Th(OH)8] 4+.” Inorganic Chemistry, vol. 46: 6804–10...\..\references\Others\Brendebach_Altmaier_Rothe_Neck_Denecke_2007_EXAFS_Study.pdf
Brush, L.H. 1990. Test Plan for Laboratory and Modeling Studies of Repository and Radionuclide Chemistry for the Waste Isolation Pilot Plant. SAND90-0266. ERMS 225053. Albuquerque: Sandia National Laboratories...\..\references\Others\Brush_1990_Test_Plan_for_Laboratory_and_Modeling_Studies_SAND90_0266.pdf
Brush, L.H. 1995. Systems Prioritization Method—Iteration 2 Baseline Position Paper: Gas Generation in the Waste Isolation Pilot Plant (March 17). ERMS 228740. Albuquerque: Sandia National Laboratories...\..\references\Others\Brush_1995_Systems_Prioritization_Method_Iteration_2_WPO28740.pdf
Brush, L.H. 2005. Results of Calculations of Actinide Solubilities for the WIPP Performance Assessment Baseline Calculations (May 18). ERMS 539800. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_2005_Results_of_Calculations_of_Actinide_Solubilities_ERMS539800.pdf
Brush, L.H., R.C. Moore, and N.A. Wall. 2001. Response to EEG-77, Plutonium Chemistry under Conditions Relevant for WIPP Performance Assessment: Review of Experimental Results and Recommendations for Future Work, by V.M. Oversby (March 15). ERMS 517373. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_Moore_Wall_2002_Response_to_EEG_77_Plutonium_Chemistry_under_Conditions_ERMS517373.pdf
Brush, L.H., and Y. Xiong. 2003a. Calculation of Actinide Solubilities for the WIPP Compliance Recertification Application(Rev. 1). AP-098. ERMS 527714. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_Xiong_2003_Calculation_of_Actinide_Solubilities_AP098_ERMS527714.pdf
Brush, L.H., and Y. Xiong. 2003b. Calculation of Organic Ligand Concentrations for the WIPP Compliance Recertification Application. ERMS 527567. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_Xiong_2003_Calculation_of_Organic_Ligand_Concentrations_for_the_WIPP_CRA_ERMS527567.pdf
Brush, L.H., and Y. Xiong. 2003c. Calculation of Organic Ligand Concentrations for the WIPP Compliance Recertification Application and for Evaluating Assumptions of Homogeneity in WIPP PA. ERMS 531488. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_Xiong_2003_Calculation_of_Organic_Ligand_Concentrations_for_WIPP_CRA_and_Evaluating_ERMS531488.pdf
Brush, L.H., and Y. Xiong, 2005a. Calculation of Organic-Ligand Concentrations for the WIPP Performance-Assessment Baseline Calculations (May 4). ERMS 539635. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_and_Xiong_2005_Calculation_of_Organic_Ligand_Concentrations_for_WIPP_PABC_ERMS539635.pdf
Brush, L.H., and Y. Xiong. 2005b. Calculation of Actinide Solubilities for the WIPP Performance-Assessment Baseline Calculations, Analysis Plan AP-120, Rev. 0(Rev. 0). AP-120. ERMS 539255. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Brush_Xiong_2005_Calculation_of_Actinide_Solubilities_for_the_WIPP_PABC_ERMS539255.pdf
Brush, L.H., and J.W. Garner. 2005. Letter to D. Kessel (Subject: Additional Justification for the Insignificant Effect of Np on the Long-Term Performance of the WIPP). 1 February 2005. ERMS 538533. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM.
Brush, L.H., Y. Xiong, J.W. Garner, A. Ismail, and G.T. Roselle. 2006. Consumption of Carbon Dioxide by Precipitation of Carbonate Minerals Resulting from Dissolution of Sulfate Minerals in the Salado Formation in Response to Microbial Sulfate Reduction in the WIPP. ERMS 544785. Carlsbad, NM: Sandia National Laboratories.../../references/Others/Brush_et_al_2006_Consumption_of_Carbon_Dioxide_by_Precipitation_ERMS544785.pdf
Bundschuh, T., R. Knopp, R. Müller, J.I. Kim, V. Neck, and Th. Fanghänel. 2000. “Application of LIBD to the Determination of the Solubility Product of Thorium(IV)-Colloids.” Radiochimica Acta, vol. 88: 625–629...\..\references\Others\Bundschuh_et_al_2000_Application_of_LIBD_for_determ_Th_colloids.pdf
Büppelmann, K., S. Magirius, Ch. Lierse, and J.I. Kim. 1986. “Radiolytic Oxidation of Am (III) to Am (V) and Pu(IV) to Pu (VI) in Saline Solution.” Journal of Less- Common Metals, vol. 122: 329–36...\..\references\Others\Buppelmann_Magirius_Lierse_Kim_1986_Radiolytic_Oxidation_of_AmIII_and_PuIV.pdf
Büppelmann, K., J.I. Kim, and Ch. Lierse. 1988. “The Redox Behavior of Pu in Saline Solutions under Radiolysis Effects.” Radiochimica Acta, vol. 44/45: 65–70...\..\references\Others\Buppelmann_Kim_Lierse_1988_Effect_of_Radiolysis_on_Pu_Redox_in_Saline_Solution.pdf
Casas, I., J. De Pablo, J. Gimenez, M.E. Torrero, J. Bruno, E. Cera, R.J. Finch, and R.C. Ewing. 1998. “The Role of pe, pH, and Carbonate on the Solubility of UO2 and Uraninite under Nominally Reducing Conditions.” Geochimica et Cosmochimica Acta, vol. 62: 2223–31...\..\references\Others\Casas_et_al_1998_The_Role_of_pe_pH_and_Carbonate.pdf
Choppin, G.R. 1983. “Solution Chemistry of the Actinides,” Radiochimica Acta, vol. 32: 43–53...\..\references\Others\Choppin_1983_Solution_Chemistry_of_Actinides.pdf
Choppin, G.R. 1988. Letter to L.H. Brush. 29 December 1988. WIPP Central Files, Tallahassee, FL...\..\references\Others\Choppin_1988_Unpublished_Letter_to_Brush_December_29_1988.pdf
Choppin, G.R., and L.F. Rao. 1992. “Reduction of Neptunium(VI) by Organic Compounds.” Transuranium Elements: A Half Century (pp. 262–75). L.R. Morss and J. Fuger, eds. Washington, DC: American Chemical Society...\..\references\Others\Choppin_Rao_1992_Reduction_of_NpVI_by_Organic_Compounds.pdf
Choppin, G.R. 1999. “Utility of Oxidation State Analogs in the Study of Plutonium Behavior.” Radiochimica Acta, vol. 85: 89–95...\..\references\Others\Choppin_1999_Utility_of_Oxidation_State_Analogs_in_Study_of_Pu_Behavior.pdf
Choppin, G.R., A.H. Bond, M. Borkowski, M.G. Bronikowski, J.F. Chen, S. Lis, J. Mizera, O. Pokrovsky, N.A. Wall, Y.X. Xia, and R.C. Moore. 2001. Waste Isolation Pilot Plant Actinide Source Term Test Program: Solubility Studies and Development of Modeling Parameters. SAND99-0943. ERMS 518556. Albuquerque: Sandia National Laboratories...\..\references\Others\Choppin_et_al_WIPP_Actinide_Source_Term_Test_Program_SAND99_0943.pdf
Choppin, G.R., J. Liljenzin, and J.O. Rydberg. 2004. Radiochemistry and Nuclear Chemistry. 3rd ed. Woburn, MA: Butterworth-Heinenmann...\..\references\Others\Choppin_Liljenzin_Rydberg_2004_Radionuclides_in_Nature.pdf
Clark, D.L., D.E. Hobart, and M.P. Neu. 1995. “Actinide Carbonate Complexes and Their Importance in Actinide Environmental Chemistry.” Chemical Reviews, vol. 95: 25...\..\references\Others\Clark_Hobart_1995_Actinide_Carbonate_Complexes_and_Their_Importance_in_Actinide.pdf
Clark, D.L., and C.D. Tait. 1996. Monthly reports under Sandia National Laboratories Contract AP2274. Sandia WIPP Central File A: WBS 1.1.10.1.1. WPO 31106...\..\references\Others\Clark_Tait_1996_Monthly_Reports_Under_SNL_Contract_AP2274.pdf
Cleveland, J.M. 1979. The Chemistry of Plutonium. La Grange Park, IL: American Nuclear Society.../../references/Others/Cleveland_1979_The_Chemistry_of_Plutonium.pdf
Cotsworth, E. 2005. Letter to I. Triay (1 Enclosure). 4 March 2005. ERMS 538858. U.S. Environmental Protection Agency, Office of Air and Radiation, Washington, DC...\..\references\Others\Cotsworth_to_Triay_March_4_2005_EPA_Letter_on_Conducting_ERMS538858.pdf
Cotton, F.A., and G. Wilkinson. 1988. Advanced Inorganic Chemistry. 5th ed. New York: Wiley...\..\references\Others\Cotton_Wilkinson_1988_Advanced_Inorganic_Chemistry.pdf
Crawford, B.A., and C.D. Leigh. 2003. Estimate of Complexing Agents in TRU Waste for the Compliance Recertification Application (August 28). ERMS 531107. Carlsbad, NM: Los Alamos National Laboratory...\..\references\Others\Crawford_Leigh_2003_Estimate_of_Complexing_Agents_in_TRU_Waste_for_CRA_ERMS531107.pdf
Cui, D., and K. Spahiu. 2002. “The Reduction of U(VI) on Corroded Iron under Anoxic Conditions.” Radiochimica Acta, vol. 90: 623–28...\..\references\Others\Cui_Spahiu_2002_Reduction_UVI_on_Corroded_Iron.pdf
Dai, M., J.M. Kelly, and K.O. Buesseler. 2002. “Sources and Migration of Plutonium in Groundwater at Savannah River Site.” Environmental Science and Technology, vol 36: 3690–99...\..\references\Others\Dai_Kelly_Buesseler_2002_Source_of_Migration_of_Pu_in_GW.pdf
Dai, M., K.O. Buesseler, and S.M. Pike. 2005. “Plutonium in Groundwater at the 100K-Area of the U.S. DOE Hanford Site.” Journal of Contaminant Hydrology, vol. 76: 167–89...\..\references\Others\Dai_Buessler_Pike_2005_Pu_in_GW_at_the_100K_Area_Of_DOE_Hanford_Site.pdf
David, F., A.G. Maslennikov, and V.P. Peretrukhin. 1990. “Electrochemical Reduction of Actinides ions in Aqueous solution: Application to Separations and Some Intermetallic Compound Synthesis.” Journal of Radioanalytical Nuclear Chemistry, vol. 143: 415–26...\..\references\Others\David_Maslennikov_Pietruchin_1990_Electrochemical_Reduction_of_Actinides_Ions_in_Aqueous_Solution.pdf
Degueldre, C., and A. Kline. 2007. “Study of Thorium Association and Surface Precipitation on Colloids.” Earth and Planetary Science Letters, vol. 264: 104–13...\..\references\Others\Degueldre_Kline_2007_Study_of_Th_Association_to_Surface_Precipitation_on_Colloids.pdf
Deng, H., S. Johnson, Y. Xiong, G.T. Roselle, and M. Nemer. 2006. Analysis of Martin Marietta MagChem 10 WTS-60 MgO (November 14). ERMS 544712. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Deng_et_al_2006_Analysis_of_Martin_Marietta_MagChem_ERMS544712.pdf
Diaz Arocas, P., and B. Grambow. 1998. “Solid-liquid Phase Equilibria of U(VI) in NaCl Solutions.” Geochimica et Cosmochimica Acta, vol. 62: 245–63...\..\references\Others\Diaz_Arocas_Grambow_1998_Solid_liquid_Phase_Equilibria.pdf
Dodge, C.J., A.J. Francis, J.B. Gillow, G.P. Halada, C. Eng, and C.R. Clayton. 2002. “Association of Uranium with Iron Oxides Typically Formed on Corroding Steel Surfaces.” Environmental Science and Technology, vol. 36: 3504–11...\..\references\Others\Dodge_Francis_Gillow_Halada_Eng_Clayton_2002_Association_U_with_Iron_Oxides.pdf
Downes, P.S. 2003a. Memorandum to L.H. Brush. Subject: Spreadsheet Calculations of Actinide Solubilities for the WIPP Compliance Recertification Application. 21 April 2003. ERMS 528395. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Downes_to_Brush_2003_Spreadsheet_Calculations_of_Actinide_Solubilities_for_CRA_ERMS528395.pdf
Downes, P.S. 2003b. Spreadsheet Calculations of Actinide Solubilities for the WIPP Compliance Recertification Application in Support of AP-098, Calculation of Actinide Solubilities for the WIPP Compliance Recertification Application, Analysis Plan AP-098, Rev. 1. ERMS 530441. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Downes_to_Brush_2003_April_21_Spreadsheet_Calculations_of_Actinide_Solubilities_ERMS530441.pdf
Draganic, I.G., and Z.D. Draganic. 1971. “Primary Products of Water Radiolysis: Oxidizing Species–the Hydroxyl Radical and Hydrogen Peroxide.” The Radiation Chemistry of Water (pp. 91–121). New York: Academic...\..\references\Others\Draganic_Draganic_1971_The_radiation_Chemistry_of_Water.pdf
Drez, P.E. 1991. “Preliminary Nonradionuclide Inventory of CH-TRU Waste,” Preliminary Comparison with 40 CFR Part 191, Subpart B for the Waste Isolation Pilot Plant, December 1991. Volume 3: Reference Data (pp. A-43 through A-53). Eds. R.P. Rechard, A.C. Peterson, J.D. Schreiber, H.J. Iuzzolino, M.S. Tierney, and J.S. Sandha. SAND91-0893/3. Albuquerque: Sandia National Laboratories, WIPP Performance Assessment Division...\..\references\Others\WIPP_PA_1991_Reference_Data_SAND91_0893_3.pdf
Ekberg, C., Y. Albinsson, M.J. Comarmond, and P.L. Brown. 2000. “Studies on the Behavior of Thorium(IV).” Journal of Solution Chemistry, vol. 29, no. 1: 63–86...\..\references\Others\Ekberg_Albinsson_Comarmond_Brown_2000_Studies_on_the_Behavior_of_ThIV.pdf
Eriksen, T.E., P. Ndamamba, D. Cui, J. Bruno, M. Caceci, and K. Spahiu. 1993. SKB Technical Report 93-18. Stockholm: Svensk Kärnbränsleforsorjning AB...\..\references\Others\Ericksen_et_al_1993_Solubility_of_Redox_Sensitive_99Tc_and_237Np.pdf
Ershov, B.G., M. Kelm, E. Janata, A.V. Gordeev, and E. Bohnert. 2002. “Radiation-Chemical Effects in the Near-Field of a Final Disposal Site: Role of Bromine on the Radiolytic Processes in NaCl-Solutions.” Radiochimica Acta, vol. 90: 617...\..\references\Others\Ershov_Kelm_Janata_Gordeev_Bohnert_2002_Radiation_Effects_Role_of_Bromine.pdf
Fanghänel, Th., V. Neck, and J.I. Kim. 1995. “Thermodynamics of Neptunium(V) in Concentrated Salt Solutions: II. Ion Interaction (Pitzer) Parameters for Np(V) Hydrolysis Species and Carbonate Complexes.” Radiochimica Acta, vol. 69: 169–76...\..\references\Others\Fanghanel_Neck_Kim_1995_Thermodynamics_of_NpV_in_Concentrated_Salt_Solutions.pdf
Fanghänel, Th., and I.J. Kim. 1998. “Spectoscopic Evaluation of Thermodynamics of Trivalent Actinides in Brines.” Journal of Alloys and Compounds, vol. 271–273: 728–737...\..\references\Others\Fanghanel_Kim_1998_Spectroscopic_Evaluation_of_Thermodynamics.pdf
Fanghänel, T., and V. Neck. 2002. “Aquatic Chemistry and Solubility Phenomena of Actinide Oxides/hydroxides.” Pure Applied Chemistry, vol. 74: 1895–1907...\..\references\Others\Fanghanel_Neck_2002_Aquatic_Chemistry_and_Solubility_Phenomena_of_An_Oxides_Hydroxides.pdf
Farrell, J., W.D. Bostick, R.J. Jarabeck, and J.N. Fiedor. 1999. “Uranium Removal from Ground Water Using Zero Valent Iron Media.” Ground Water, vol. 37: 618–24...\..\references\Others\Farrell_Bostick_Jarabeck_Fiedor_1999_U_removal_from_Ground_Water.pdf
Felmy, A.R., D. Rai, J.AS. Schramke, and J.L. Ryan. 1989. “The Solubility of Plutonium Hydroxide in Dilute Solution and in High-Ionic Strength Chloride Brines.” Radiochimica Acta, vol. 48: 29–35...\..\references\Others\Felmy_et_al_1989_Solubility_of_Pu_Hydroxide.pdf
Felmy, A.R., D. Rai, and R.W. Fulton. 1990. “The Solubility of AmOHCO3(cr) and the Aqueous Thermodynamics of the System Na+-Am3+-HCO3--CO 32--OH—H2O.” Radiochim. Acta, vol. 50: 193–204...\..\references\Others\Felmy_Rai_Fulton_1990_Solubility_of_AmOHCO.pdf
Felmy, A.R., M.J. Mason, and D. Rai, 1991. “The Solubility of Hydrous Thorium(IV) Oxide in Chloride Media: Development of an Aqueous Ion-Interaction Model.” Radiochimica Acta, vol. 55: 177–85...\..\references\Others\Felmy_Mason_Rai_1991_Solubility_of_Hydrous_ThIV_Oxide_in_Chloride_Media.pdf
Felmy, A.R., D. Rai, S.M. Sterner, M.J. Mason, N.J. Hess, and S.D. Conradson. 1996. Thermodynamic Models for Highly Charged Aqueous Species: The Solubility of Th(IV) Hydrous Oxide in Concentrated NaHCO3 and Na2CO3 Solutions (August 14). ERMS 240226. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Felmy_et_al_1996_Thermodynamic_Models_for_Highly_Charged_Aqueous_Species_WPO40226.pdf
Fiedor, J.N., W.D. Bostick, R.J. Jarabek, and J. Farrell. 1998. “Understanding the Mechanism of Uranium Removal from Groundwater by Zero-Valent Iron Using X-Ray Photoelectron Spectroscopy.” Environmental Science and Technology, vol. 32: 1466–73...\..\references\Others\Fiedor_Bostick_Jarabeck_Farrell_1998_Understanding_the_Mechanism.pdf
Francis, A.J. 1990. “Microbial Dissolution and Stabilization of Toxic Metals and Radionuclides in Mixed Wastes.” Experentia, vol. 46: 840–51...\..\references\Others\Francis_1990_Microbial_Dissolution_and_Stabilz_of_Toxic_Metals.pdf
Francis, A.J. 1998. “Biotransformation of Uranium and Other Actinides in Radioactive Wastes.” Journal of Alloys and Compounds, vol. 271–273: 78–84...\..\references\Others\Francis_1998_Biotransformation_of_U_and_other_An_in_Radioactive_Wastes.pdf
Francis, A.J., and J.B. Gillow. 1994. Effects of Microbial Processes on Gas Generation Under Expected Waste Isolation Pilot Plant Repository Conditions. Progress Report through 1992. SAND93-7036. Albuquerque: Sandia National Laboratories...\..\references\Others\Francis_Gillow_1994_Effects_of_Microbial_Processes_on_Gas_Generation_SAND93_7036.pdf
Francis, A.J., C.J. Dodge, J.B. Gillow, and H.W. Papenguth. 2000. “Biotransformation of Uranium Compounds in High Ionic Strength Brine by a Halophilic Bacterium under Denitrifying Conditions.” Environmental Science and Technology, vol. 34: 2311...\..\references\Others\Francis_Dodge_Gillow_Papenguth_2000_Biotransformation_of_U_by_Halophilic_Bacterium.pdf
Francis, A.J., C.J. Dodge, and J. B. Gillow. 2008. “Reductive Dissolution of Pu(IV) by Costridium sp. under Anaerobic Conditions.” Environmental Science and Technology., vol. 42: 2355–60...\..\references\Others\Francis_Dodge_Gillow_2008_Reductive_dissolution_of_PuIV.pdf
Fredrickson, J.K., J.M. Zachara, D.W. Kennedy, M.C. Duff, Y.A. Gorby, S.W. Li, and K.M. Krupka. 2000. “Reduction of U(VI) in Goethite (α-FeOOH) Suspensions by a Dissimilatory Metal-Reducing Bacteria.” Geochimica et. Cosmochimica Acta, vol. 64: 3085–98...\..\references\Others\Fredrickson_et_al_2000_Reduction_of_UVI_in_Geothite.pdf
Garner, J.W. 1996. Radioisotopes to be Used in the 1996 CCA Calculations. ERMS 540572. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Garner_to_Stockman_1996_March_15_Radioisotopes_to_be_Used_in_CCA_Calculations_WPO35202.pdf
Garner, J., and C. Leigh. 2005. Analysis Package for PANEL: CRA-2004 Performance Assessment Baseline Calculation(Revision 0). ERMS 540572. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Garner_Leigh_2005_Analysis_Package_for_PANEL_CRA_2004_PABC_ERMS540572.pdf
Gayer, K.H., and H. Leider. 1957. “The Solubility of Uranium (IV) Hydroxide in Solutions of Sodium Hydroxide and Perchloric Acid at 25°C.” Canadian Journal of Chemistry, vol. 35: 5–7...\..\references\Others\Gayer_Leider_1957_The_Solubility_of_UIV_Hydroxide.pdf
Giambalvo, E.R. 2002a. Memorandum to L.H. Brush (Subject: Recommended Parameter Values for Modeling An(III) Solubility in WIPP Brines). 25 July 2002. ERMS 522982. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2002_July_25_Recommended_Parameter_Values_for_Modeling_AnIII_Solubility_ERMS522982.pdf
Giambalvo, E.R. 2002b. Memorandum to L.H. Brush (Subject: Recommended Parameter Values for Modeling Organic Ligands in WIPP Brines). 25 July 2002. ERMS 522981. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2002_July_25_Recommended_Parameter_Values_for_Modeling_Organic_Ligands_ERMS522981.pdf
Giambalvo, E.R. 2002c. Memorandum to L.H. Brush (Subject: Recommended Parameter Values for Modeling An(IV) Solubility in WIPP Brines). 26 July 2002. ERMS 522986. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2002_July_26_Recommended_Parameter_Values_for_Modeling_AnIV_Solubility_ERMS522986.pdf
Giambalvo, E.R. 2002d. Memorandum to L.H. Brush (Subject: Recommended Parameter Values for Modeling An(V) Solubility in WIPP Brines). 26 July 2002. ERMS 522990. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2002_July_26_Recommended_Parameter_Values_for_Modeling_AnV_Solubility_ERMS522990.pdf
Giambalvo, E.R. 2002e. Memorandum to L.H. Brush (Subject: Recommended m0/RT Values for Modeling the Solubility of Oxalate Solids in WIPP Brines). 31 July 2002. ERMS 523057. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2002_July_31_Recommended_Normalized_Chemical_Potential_Values_ERMS523057.pdf
Giambalvo, E.R., 2003. Memorandum to L.H. Brush (Subject: Release of FMT Database FMT_021120.CHEMDAT). 10 March 2003. ERMS 526372. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Giambalvo_to_Brush_2003_March_10_Release_of_FMT_Database_FMT_021120_ERMS526372.pdf
Gillow, J.B., M. Dunn, A.J. Francis, D.A. Lucero, and H.W. Papenguth. 2000. “The Potential of Subterranean Microbes in Facilitating Actinide Migration at the Grimsel Test Site and Waste Isolation Pilot Plant,” Radiochemica Acta, vol. 88: 769–774...\..\references\Others\Gillow_et_al_2000_The_Potential_Microbes_Facilit_Transport.pdf
Grambow, B., E. Smailos, H. Geckeis, R. Muller, and H. Hentschel. 1996. “Sorption and Reduction of Uranium (VI) on Iron Corrosion Products under Reducing Saline Conditions.” Radiochimica Acta, vol. 74: 149–54...\..\references\Others\Grambow_et_al_1996_Sorption_and_Reduction_of_UVI_on_Fe_Corrossion_Prod.pdf
Grenthe, I., D. Ferri, F. Salvatore, and G. Riccio. 1984. “Studies on Metal Carbonate Equilibria. Part 10. A Solubility Study of the Complex Formation in the Uranium (VI)-Water-Carbon Dioxide (g) System at 25 ºC” Journal of Chemical Society, Dalton Trans.: 2439–43...\..\references\Others\Grenthe_Ferri_Salvatore_and_Riccio_1984_Complex_Formation_in_UVI_Water_CO2_System.pdf
Gu, B., L. Liang, M.J. Dickey, X. Yin, and S. Dai. 1998. “Reductive Precipitation of Uranium (VI) by Zero-Valent Iron.” Environmental Science and Technology, vol. 32: 3366–73...\..\references\Others\Gu_Liang_Dickey_Yin_Dai_1998_Reductive_Precipitation_of_UVI_by_Fe0.pdf
Guillaumont, R., T. Fanghänel, J. Fuger, I. Grenthe, V. Neck, D.A. Palmer, and M.H. Rand. 2003. “Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium.” F.I. Mompean, M. Illemassene, C. Domenech-Orti, and K. Ben Said, eds. Chemical Thermodynamics. Vol. 5. Amsterdam: Elsevier...\..\references\Others\Guillaumont_et_al_2003_Update_on_Chemical_Thermodynamics.pdf
Harvie, C.E., N. Møller, and J.H. Weare. 1984. “The Prediction of Mineral Solubilities in Natural Waters, The Na-K-Mg-Ca-H-Cl-SO4-OH-HCO3-CO2-H 2O System to High Ionic Strengths at 25°C.” Geochimica et Cosmochimica Acta, vol. 48: 723–51...\..\references\Others\Harvie_Moller_Weare_1984_Mineral_Solubilities_in_Natural_Waters.pdf
Haschke, J.M., and T.E. Ricketts. 1995. Plutonium Dioxide Storage: Conditions for Preparing and Handling. LA-12999. Los Alamos, NM: Los Alamos National Laboratory...\..\references\Others\Haschke_Ricketts_1995_Pu_Dioxide_Storage_Conditions_for_Preparing_and_Handling.pdf
Haschke, J.M., T.H. Allen, and L.A. Morales. 2000. “Reaction of Plutonium Dioxide with Water: Formation and Properties of PuO2+x.” Science, vol. 287: 285–87...\..\references\Others\Haschke_Allen_Morales_2000_Reaction_PuO2_with_Water.pdf
Hobart, D.E., K. Samhoun, and J. R. Peterson. 1982. “Spectroelectrochemical Studies of the Actinides: Stabilization of Americium (IV) in Aqueous Carbonate Solution.” Radiochimica Acta, vol. 31: 139–45...\..\references\Others\Hobart_Samhoun_Peterson_1982_Stabilization_of_AmIV_in_Carbonate_Solution.pdf
Hobart, D.E., 1990. “Actinides in the Environment.” Proceedings of the Robert A. Welch Foundation Conference on Chemical Research, No. XXXIV: 50 Years With Transuranium Elements (pp. 378–436). Houston, TX: Robert A. Welch Foundation...\..\references\Others\Hobart_1990_Actinides_in_Environment_pp_378_436.pdf
Hobart, D.E., and R.C. Moore. 1996. Analysis of Uranium (VI) Solubility Data for WIPP Performance Assessment. (May 28, 1996). AP-028. Unpublished report. Albuquerque: Sandia National Laboratories...\..\references\Others\Hobart_Moore_1996_Analysis_of_UV1_Solubility_Data.pdf
Huang, F.Y C., P.V. Brady, E.R. Lindgren, and P. Guerra. 1998. “Biodegradation of Uranium-Citrate Complexes: Implications for Extraction of Uranium from Soils.” Environmental Science and Technology, vol. 32: 379...\..\references\Others\Huang_et_al_1998_Biodegradation_of_U_citrate_complexes.pdf
Icopini, G.A., J. Boukhalfa, and M.P. Neu. 2007. “Biological Reduction of Np(V) and Np(V) Citrate by Metal-Reducing Bacteria.” Environmental, Science, & Technology, vol. 41: 2764–69...\..\references\Others\Neu_2007_Biological_Reduction_of_NpV_by_Metal_Reducing_Bacteria.pdf
Ionova, G., C. Madic, and R. Guillamount. 1998. “About the Existence of Th(III) in Aqueous Solution.” Polyhedron, vol. 17: 1991–95...\..\references\Others\Ionova_Madic_Guillamount_1998_About_Existence_of_ThIII.pdf
Itagaki, H., S. Nakayama, S. Tanaka, and M. Yamawaki. 1992. “Effect of Ionic Strength on the Solubility of Neptunium(V) Hydroxide.” Radiochimica Acta, vol. 58/59: 61–66...\..\references\Others\Itagaki_et_al_1992_Effect_of_Ionic_Strength_on_NpV_Hydroxide_Solubility.pdf
Johnson, G.L., and L.M. Toth. 1978. Plutonium(IV) and Thorium(IV) Hydrous Polymer Chemistry. ORNL/TM-6365. Oak Ridge, TN: Oak Ridge National Laboratory, Chemistry Division...\..\references\Others\Johnson_Toth_1978_PlutoniumIV_and_ThoriumIV_Hydrous_Polymer_ORNL_TM_6365.pdf
Katz, J.J., G.T. Seaborg, and L.R. Morss. 1986. The Chemistry of the Actinide Elements. 2nd ed. New York: Chapman and Hall...\..\references\Others\Katz_Seaborg_Morss_1986_The_Chemistry_of_Actinide_Elements.pdf
Keller, C. 1971. The Chemistry of Transuranium Elements. Weinheim, Germany: Verlag Chemie...\..\references\Others\Keller_1971_The_Chemistry_of_Transuranium_Elements.pdf
Kelm, M., I. Pashalidis, and J.I. Kim. 1999. “Spectroscopic Investigation on the Formation of Hypochlorite by Alpha Radiolysis in Concentrated NaCl Solutions.” Applied Radiation and Isotopes, vol. 51: 637–42...\..\references\Others\Kelm_Pashalidis_Kim_1999_Formation_of_Hypochlorite_by_Alfa_Radiolysis.pdf
Kersting, A.B., D.W. Efurd, D.L. Finnegan, D.J. Rokop, D.K. Smith, and J.L. Thompson. 1999. “Migration of Plutonium in Grand Water at the Nevada Test Site.” Nature, vol. 397: 56–59...\..\references\Others\Kersting_et_al_1999_Pu_Migration_in_GW.pdf
Khasanova, A.B., N.S. Shcherbina, S.N. Kalmykov, Yu.A. Teterin, and A.P. Novikov. 2007. “Sorption of Np(V), Pu(V), and Pu(VI) on Colloids of Fe(III) Oxides and Hydrous Oxides and MnO2.” Radiochemistry, vol. 49: 419–25...\..\references\Others\Khasanova_et_al_2007_Sorption_of_NpV_and_PuIV_on_Colloids.pdf
Kim, J.I., M. Bernkopf, Ch. Lierse, and F. Koppold. 1984. “Hydrolysis Reactions of Am(III) and Pu(VI) Ions in Near-Neutral Solutions.” Geochemical Behavior of Disposed Radioactive Waste (pp. 115–34). G.S. Barney, J.D. Navratill, and W.W. Schultz, eds. ACS Symposium Series No. 246. Washington, DC: American Chemical Society...\..\references\Others\Kim_et_al_1984_Hydrolysis_AmIII_and_PuVI_in_Near_Neutral_Solutions.pdf
Kim, J.I., Ch. Apostolidis, G. Buckau, K. Buppelmann, B. Kanellakopulos, Ch. Lierse, S. Magirius, R. Stumpe, I. Hedler, Ch. Rahner, and W. Stoewer. 1985. Chemisches Verhalten von Np, Pu und Am in verschiedenen konzentrierten Salzloesunger = Chemical Behaviour of Np, Pu, and Am in Various Brine Solutions. RCM 01085. Munich, Germany: Institut für Radiochemie der Technische Universitaet Muenchen. (Available from National Technical Information Service, 555 Port Royal Road, Springfield, VA 22161, 703/487-4650 as DE857 2334.)..\..\references\Others\Kim_et_al_1985_Chemical_Behaviour_of_Np_Pu_and_Am_RCM_01085.pdf
Kim, J.I. 1991. “Actinide Colloid Generation in Groundwater.” Radiochimica Acta, vol. 52/53: 71–81...\..\references\Others\Kim_1991_An_Colloid_Generation_in_Groundwater.pdf
Kim, W.H., K.C. Choi, K.K. Park, and T.Y. Eom. 1994. “Effects of Hypochlorite Ion on the Solubility of Amorphous Schoepite at 25°C in Neutral to Alkaline Aqueous Solutions.” Radiochimica Acta, vol. 66/67: 45–49...\..\references\Others\Kim_Choi_Park_Eom_1994_Effects_of_Hypochlorite_Ion_on_the_Schoepite_Solubility.pdf
Klapötke, T.M., and A. Schulz. 1997. “The First Observation of a Th3+ ion in Aqueous Solution.” Polyhedron, vol. 16: 989–91...\..\references\Others\Klapotke_Schultz_1997_The_First_Observation_of_a_Th_3_Ion.pdf
Korpusov, G.V., E.N. Patrusheva, and M.S. Dolidze. 1975. “The Study of Extraction Systems and the Method of Separation of Trivalent Transuranium Elements Cm, Bk, and Cf.” Soviet Radiochemistry, vol. 17: 230–36...\..\references\Others\Korpusov_Patrusheva_Dolidze_1975_The_Study_of_Extration.pdf
Kramer-Schnabel, U., H. Bischoff, R.H. Xi, and G. Marx. 1992. “Solubility Products and Complex Formation Equilibria in the Systems Uranyl Hydroxide and Uranyl Carbonate at 25°C and I=0.1M.” Radiochimica Acta, vol. 56: 183–88...\..\references\Others\Kramer_et_al_1992_Solubility_Products_and_Complex_Formation.pdf
Langmuir, D., and J.S. Herman. 1980. “Mobility of Thorium in Natural Waters at Low Temperatures.” Geochimica et. Cosmochimica Acta, vol. 44: 1753–66...\..\references\Others\Langmuir_Herman_1980_Mobility_of_Thorium_in_Natural_Waters_at_low_Temperatures.pdf
Larson, K.L.. 1996. Memorandum to R. V. Bynum. Subject: Brine Waste Contact Volumes for Scoping Analysis of Organic Ligand Concentration. 13 March 1996. ERMS 236044...\..\references\Others\Larson_to_Bynum_1996_March_13_Brine_Waste_Contact_Volumes_WPO36044.pdf
LaVerne, J.A., and L. Tandon. 2002. “H2 Production in the Radiolysis of Water on CeO2 and ZrO2.” Journal of Physical Chemistry B, vol. 106: 380–86...\..\references\Others\LaVerne_Tandon_2002_H2_Production_in_Radiolysis_of_Water_Adsorbed_on_CeO2_and_ZrO2.pdf
Leigh, C., J. Trone, and B. Fox. 2005. TRU Waste Inventory for the 2004 Compliance Recertification Application Performance Assessment Baseline Calculation(Revision 0). ERMS 541118. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Leigh_Trone_Fox_2005_TRU_Waste_Inventory_for_CRA2004_PABC_ERMS541118.pdf
Leigh, C., J. Kanney, L. Brush, J. Garner, G. Kirkes, T. Lowry, M. Nemer, J. Stein, E. Vugrin, S. Wagner, and T. Kirchner. 2005. 2004 Compliance Recertification Application Performance Assessment Baseline Calculation(Revision 0). ERMS 541521. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Leigh_et_al_2004_CRA_PABC_rev0_ERMS541521.pdf
Lemire, R.J., J. Fuger, H. Nitsche, P. Potter, M.H. Rand, J. Rydberg, K. Spahiu, J.C. Sullivan, W.J. Ullman, P. Vitorge, and H. Wanner. 2001. Chemical Thermodynamics of Neptunium and Plutonium. Amsterdam: Elsevier...\..\references\Others\Lemire_et_al_2001_Chemical_Thermodynamics_of_Np_and_Pu.pdf
Lieser, K.H., R. Hill, U. Mühlenweg, R.N. Singh, T. Shu-De, and Th. Steinkopff. 1991. “Actinides in the Environment.” Journal of Radioanalytical and Nuclear Chemistry, vol. 147: 117–31...\..\references\Others\Lieser_et_al_1991_Actinides_in_the_Environment.pdf
Lin, M.R., P. Paviet-Hartmann, Y. Xu, and W.H. Runde. 1998. “Uranyl Compounds in NaCl Solutions: Structure, Solubility and Thermodynamics.” 216th ACS National Meeting: Preprints of Extended Abstracts, vol. 38: 208. Abstract for the National Meeting of the American Chemical Society, Division of Environmental Chemistry, Boston: 23-27 August...\..\references\Others\Lin_et_al_1998_Uranyl_Compounds.pdf
Lloyd, J.R., P. Yong, and L.E. Macaskie. 2000. “Biological Reduction and Removal of Np(V) by Two Microorganisms.” Environmental Science and Technology, vol. 34: 1297–1301...\..\references\Others\Lloyd_Yong_Macaskie_2000_Bilogical_reduction_and_removal_of_NpV.pdf
LoPresti, V., S.D. Conradson, and D.L. Clark. 2007. “XANES Identification of Plutonium Speciation in RFETSl Samples.” Journal of Alloys and Compounds, vol. 444–445: 540–43...\..\references\Others\LoPresti_Conradson_and_Clark_2007_XANES_Idenfication_of_Pu_Species_in_Rocky_Flat_Soil_Samples.pdf
Lovley, D.R., E.J.P. Phillips, Y.A. Gorbi, and E.R. Landa. 1991. “Microbial Reduction of Uranium.” Nature, vol. 350: 413...\..\references\Others\Lovley_et_al_1991_Microbial_Reduction_of_Uranium.pdf
Lovley, D.R., E.E. Roden, E.J.P. Phillips, and J.C. Woodward. 1993. “Enzymatic Iron and Uranium Reduction by Sulfate-Reducing Bacteria.” Marine Geology, vol. 113: 41...\..\references\Others\Lovley_et_al_1993_Enzymatic_Iron_and_Uranium_Reduction_by_Sulfate_Reducing_Bacteria.pdf
Lucchini, J.F., M. Borkowski, M.K. Richmann, S. Ballard, and D.T. Reed. 2007. “Solubility of Nd3+ and UO22+ in WIPP Brine as Oxidation-State Invariant Analogs for Plutonium.” Journal of Alloys and Compounds, vol. 444/445: 506–11...\..\references\Others\Lucchini_et_al_2007_NdIII_and_UVI_Solubility.pdf
Lucchini, J.F., H. Khaing, M. Borkowski, M.K. Richmann, and D.T. Reed. 2008. Actinide (VI) Solubility in Carbonate-free WIPP Brine: Data Summary and Recommendations. LCO-ACP-10, LANL\ACRSP Report. Los Alamos, NM: Los Alamos National Laboratory...\..\references\Others\LCO_ACP_10.pdf
Magirus, S., W.T. Carnall, and J.I. Kim. 1985. “Radiolytic Oxidation of Am(III) to Am(V) in NaCl Solution.” Radiochimical Acta, vol. 38: 29–32...\..\references\Others\Magirius_Carnall_Kim_1985_Radiolytic_Oxidation_AmIII_to_AmV_in_NaCl_Solution.pdf
Marinot, L., and J. Fuger. 1985. “The Actinides.” Standard Potentials in Aqueous Solution (pp. 631–674). A.J. Bard, R. Parsons, and J. Jordan, eds. New York: Dekker...\..\references\Others\Marinot_Fuger_1985_The_Actinides.pdf
Meinrath, G., and J.I. Kim. 1991. “The Carbonate Complexation of the Am(III) Ion.” Radiochimica Acta, vol. 52/53: 29–34...\..\references\Others\Meinrath_Kim_1991_Carbonate_Complexation_of_AmIII_Ion.pdf
Meinrath, G., and T. Kimura. 1993. “Carbonate Complexation of the Uranyl (VI) Ion.” Journal of Alloys and Compounds, vol. 202: 89–93...\..\references\Others\Meinrath_Kimura_1993_Carbonate_Complexation_of_the_Uranyl_VI_Ion.pdf
Meyer, D., S. Fouchard, E. Simoni, and C. DenAuwer. 2002. “Selective Dissolution of Am in Basic Media in the Presence of Ferricyanide Ions: a Mechanistic and Structural Study on Am(V) and Am(VI) Compounds.” Radiochimica Acta, vol. 90: 253–58...\..\references\Others\Meyer_Fouchard_Simoni_DenAuwer_2002_Selective_Dissolution_of_Am_in_Basic_Media_with_Ferricyanide.pdf
Molecke, M.A. 1983. A Comparison of Brines Relevant to Nuclear Waste Experimentation. SAND83-0516. Albuquerque: Sandia National Laboratories...\..\references\Others\Molecke_1983_A_Comparison_of_Brines_Relevant_to_Nuclear_Waste_Experimentation_SAND83_0516.pdf
Moriyama, H., T. Sasaki, T. Kobayashi, and I. Takagi. 2005. “Systemics of Hydrolysis Constants of Tetravalent Actinide Ions.” Journal of Nuclear Science and Technology, vol. 42-7: 626–35...\..\references\Others\Moriyama_Sasaki_Kobayashi_Takagi_2005_Systematics_of_AnIV_Hydrolysis_Constants.pdf
Morss, L.R., N. Edelstein, and J. Fuger, 2006. The Chemistry of the Actinide and Transactinide Elements. 3rd ed. New York: Springer...\..\references\Others\Morss_2006_Chemistry_of_Actinide.pdf
Moulin, C., B. Amekraza, S. Hubert, and V. Moulin. 2001. “Study of Thorium Hydrolysis Species by Electrospray-Ionization Mass Spectrometry.” Analytica Chimica Acta, vol. 441: 269–79...\..\references\Others\Moulin_Amekraza_Hubert_Moulin_2001_Study_of_Th_Hydrolysis_Species_by_Electrospray.pdf
Munson, D.E., R.L. Jones, D.L. Hoag, and J.R. Ball. 1987. Heated Axisymmetric Pillar Test (Room H): In Situ Data Report (February, 1985–April, 1987), Waste Isolation Pilot Plant (WIPP) Thermal/Structural Interactions Program. SAND87-2488. Albuquerque: Sandia National Laboratories...\..\references\Others\Munson_Jones_Hoag_Ball_1987_Heated_Axisymmetric_Pillar_Test_SAND87_2488.pdf
Nakata, K., S. Nagasaki, S. Tanaka, Y. Sakamoto, T. Tanaka, and H. Ogawa. 2004. “Reduction Rate of Neptunium(V) in Heterogeneous Solution with Magnetite.” Radiochimica Acta, vol. 92: 145–49...\..\references\Others\Nakata_et_al_2004_Rduction_Rate_of_Np_Vin_Heterogeneous_Solution_with_Magnetite.pdf
Neck, V., J.I. Kim, and B. Kanellakopulos. 1992. “Solubility and Hydrolysis Behavior of Neptunium (V).” Radiochimica Acta, vol. 56:, 25–30...\..\references\Others\Neck_Kim_Kanellakopulos_1992_Solubility_and_Hydrolysis_of_NpV.pdf
Neck, V., W. Runde, and J.I. Kim. 1995. “Solid-Liquid Equilibria of Np(V) in Carbonate Solutions at Different Ionic Strengths: II.” Journal of Alloys and Compounds, vol. 225: 295–302...\..\references\Others\Neck_Runde_Kim_1995_Solid_Liquid_Equilibria_of_NpV.pdf
Neck, V., and J.I. Kim. 2001. “Solubility and Hydrolysis of Tetravalent Actinides.” Radiochimica Acta, vol. 89: 1–16...\..\references\Others\Neck_Kim_2001_Solubility_and_Hydrolysis_of_AnIV.pdf
Neck, V., R. Müller., M. Bouby, M. Altmaier, J. Rothe, M.A. Denecke, and J.I. Kim. 2002. “Solubility of Amorphous Th(IV) Hydroxide: Application of LIBD to Determine the Solubility Product and EXAFS for Aqueous Speciation.” Radiochimica Acta, vol. 90: 485–94...\..\references\Others\Neck_Altmaier_Muller_Bauer_Fanghanel_Kim_2003_Solubility_of_Crystalline_ThO2.pdf
Neck, V., M. Altmaier, R. Müller, A. Bauer, Th. Fanghänel, and J.I. Kim. 2003. “Solubility of Crystalline Thorium Dioxide.” Radiochimica Acta, vol. 91: 253–62...\..\references\Others\Neck_et_al_2003_Solubility_of_Crystalline_ThO2.pdf
Neck, V., M. Altmaier, and Th. Fanghänel. 2006. “Ion Interaction (SIT) Coefficients for the Th4+ Ion and Trace Activity Coefficients in NaClO4, NaNO3 and NaCl Solution Determined by Solvent Extraction with TBP.” Radiochimica Acta, vol. 94: 501–07...\..\references\Others\Neck_Altmaier_Fanghanel_2006_Ion_Interaction_Coefficients.pdf
Nemer, M., and J. Stein. 2005. Analysis Package for BRAGFLO: 2004 Compliance Recertification Application Performance Assessment Baseline Calculation. ERMS 540527. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Nemer_and_Stein_2005_Analysis_Package_for_BRAGFLO_ERMS540527.pdf
Nemer, M., J. Stein, and W. Zelinski. 2005. Analysis Report for BRAGFLO Preliminary Modeling Results With New Gas Generation Rates Based on Recent Experimental Results. ERMS 539437. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Nemer_Stein_Zelinski_2005_Analysis_Report_of_BRAGFLO_ERMS539437.pdf
Neretnieks, I. 1982. The Movement of a Redox Front Downstream From a Repository for Nuclear Waste. KBS Report TR 82-16. Stockholm: Svensk Kärnbränsleforsorjning AB...\..\references\Others\KBS_Report_TR_82_16.pdf
Neu, M.P., C.E. Ruggiero, and A.J. Francis. 2002. “Bioinorganic Chemistry of Plutonium and Interactions of Plutonium with Microorganisms in Plants.” Advances in Plutonium Chemistry (pp. 1967-2000). La Grange Park, IL: American Nuclear Society...\..\references\Others\Neu_Ruggiero_Francis_2002_Bioinorganic_Chemistry_of_Pu_LA_UR01_2708.pdf
National Institute of Standards and Technology (NIST). 2004. “Critical Stability Constants.” Standard Reference Database 46 (Version 8.0)...\..\references\Others\NIST_2004_Critical_Stability_Constants.pdf
Nitsche, H., K. Roberts, R.C. Gatti, T. Prussin, K. Becraft, S.C. Leung, S.A. Carpenter, and C.F. Novak. 1992. Plutonium Solubility and Speciation Studies in a Simulant of Air Intake Shaft Water from the Culebra Dolomite at the Waste Isolation Pilot Plant. SAND92-0659. WPO 23480. Albuquerque: Sandia National Laboratories...\..\references\Others\Nitsche_et_al_1992_Plutonium_Solubility_and_Speciation_Studies_SAND92_0659.pdf
Nitsche, H., K. Roberts, R. Xi, T. Prussin, K. Becraft, I. Al Mahamid, H.B. Silber, S.A. Carpenter, R.C. Gatti, and C.F. Novak. 1994. “Long-Term Plutonium Solubility and Speciation Studies in a Synthetic Brine.” Radiochimica Acta, vol. 66/67: 3–8...\..\references\Others\Nitsche_et_al_1994_Long_Term_Plutonium.pdf
Nitsche, H., K. Roberts, K. Becraft, T. Prussin, D. Keeney, S. Carpenter, and D. Hobart. 1995. Solubility and Speciation Results from Over- and Under-saturation Experiments on Neptunium, Plutonium and Americium in water from Yucca Mountain Region Well UE-25p#1. Report LA-13017-MS. Los Alamos, NM: Los Alamos National Laboratory...\..\references\Others\Nitsche_et_al_Solubility_and_Speciation_Results_Report_LA13017_MS.pdf
Novak, C.F. 1995. The WIPP Actinide Source Term: Test Plan for the Conceptual Model and the Dissolved Concentration Submodel. SAND95-1985. WPO 27860. Albuquerque: Sandia National Laboratories...\..\references\Others\Novak_1995_WIPP_Actinide_Source_Term_Test_Plan_SAND95_1895.pdf
Novak, C.F., R.C. Moore, and R.V. Bynum. 1996. “Prediction of Dissolved Actinide Concentrations in Concentrated Electrolyte Solutions: A Conceptual Model and Model Results for the Waste Isolation Pilot Plant (WIPP).” Proceedings of the International Conference on Deep Geological Disposal of Radioactive Waste. Winnipeg, Manitoba: 16-19 September 1996...\..\references\Others\Novak_Moore_Bynum_1996_Prediction_of_Dissolved_Actinide.pdf
Novak, C.F., and R.C. Moore. 1996. Sandia National Laboratories Technical Memo. Subject: Estimates of Dissolved Concentrations for III, IV, V, and VI Actinides in Salado and Castile Brine Under Anticipated Repository Conditions. 28 March 1996. WIPP Central File A: WBS 1.2.0.7.1; WBS 1.1.10.1.1: WPO 36207. ..\..\references\Others\Novak_Moore_to_Siegel_1996_March_28_Estimates_of_Dissolved_Concentrations_WPO36207.pdf
Novikov, A.P., S.N. Kalmykov, S. Utsunomiya, C. Ewing, F. Horreard, A. Merkulov, S.E. Clark, V.V. Tkachev, and B.F. Myasoedov. 2006. “Colloid Transport of Plutonium in the Far-Field of the Mayak Production Association, Russia.” Science, vol. 314: 638–41...\..\references\Others\Novikov_et_al_2006_Pu_Colloid_Transport_in_the_Far_Field.pdf
O’Loughlin, E.J., S.D. Kelly, R.E. Cook, R. Csencsits, and K.M. Kemner. 2003. “Reduction of Uranium (VI) by Mixed Iron (II)/ Iron (III) Hydroxide (Green Rust): Formation of UO2 Nanoparticles.” Environmental Science and Technology, vol. 37: 721–27...\..\references\Others\O_Loughlin_et_al_2003_Reduction_UVI_by_FeII_and_FeIII.pdf
Okajima, S., and D.T. Reed. 1993. “Initial Hydrolysis of Pu(VI).” Radiochimica Acta, vol. 60: 173–84...\..\references\Others\Okajima_Reed_1993_Initial_Hydrolysis_of_PlutoniumVI.pdf
Okamoto, Y., Y. Mochizuki, and S. Tsushim. 2003. “Theoretical Study of Hydrolysis Reactions of Tetravalent Thorium Ion.” Chemical Physics Letters, vol. 373: 213–17...\..\references\Others\Okamoto_Mochizuki_Tsushim_2003_Theoretical_Study_of_Hydrolysis_Reactions.pdf
Orlandini, J.A., W.R. Penrose, and D.M. Nelson. 1986. “Pu(V) as the Stable form of Oxidized Plutonium in Natural Waters.” Marine Chemistry, vol. 18: 49–57...\..\references\Others\Orlandini_Penrose_Nelson_1986_PuV_as_the_Stable_Form_of_Plutonium.pdf
Östhols, E., J. Bruno, and I. Grenthe. 1994. “On the Influence of Carbonate on Mineral Dissolution: III. The Solubility of Microcrystalline ThO2 in CO2-H2O Media.” Geochimica et Cosmochimica Acta, vol. 58: 613–623...\..\references\Others\Osthol_Bruno_Grenthe_1994_On_the_Influence_of_Carbonite.pdf
Oversby, V.M. 2000. Plutonium Chemistry under Conditions Relevant for WIPP Performance Assessment: Review of Experimental Results and Recommendations for Future Work. (September). EEG-77. Albuquerque: Environmental Evaluation Group...\..\references\Others\Oversby_2000_Plutonium_Chemistry_under_Conditions_Relevant_for_WIPP_PA_EEG77.pdf
Papenguth, H.W., and Y.K. Behl. 1996. Test Plan for Evaluation of Colloid-Facilitated Actinide Transport at the WIPP. TP 96-01. ERMS 231337. Albuquerque: Sandia National Laboratories...\..\references\Others\Papenguth_Behl_1996_Test_Plan_for_Evaluation_of_Colloid_Facilitated_Actinide_ERMS417319.pdf
Papenguth, H.W. 1996a. Letter to Chrstine T. Stockman (Subject: Parameter Record Package for Colloidal Actinide Source Term Parameters, Attachment A: Rationale for Definition of Parameter Values for Microbes). 7 May 1996. ERMS 235856. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Papenguth_tp_Stockman_1996_May_7_Microbes_ERMS235856.pdf
Papenguth, H.W. 1996b. Letter to Christine T. Stockman (Subject: Parameter Record Package for Colloidal Actinide Source Term Parameters, Attachment A: Rationale for Definition of Parameter Values for Humic Substances). 7 May 1996. ERMS 235855. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Papenguth_to_Stockman_1996_May_7_Humic_Substances_ERMS235855.pdf
Papenguth, H.W. 1996c. Letter to Christine T. Stockman (Subject: Parameter Record Package for Colloidal Actinide Source Term Parameters, Attachment A: Rationale for Definition of Parameter Values for Mineral Fragment Type Colloids). 7 May 1996. ERMS 235850. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Papenguth_to_Stockman_1996_May_7_Mineral_Fragment_Type_ERMS235850.pdf
Pashalidis, I., J.I. Kim, Ch. Lierse, and J. Sullivan. 1993. “The Chemistry of Pu in Concentrated Aqueous NaCl Solution: Effects of Alpha Self-Radiolysis and the Interaction Between Hypochlorite and Dioxoplutonium (VI).” Radiochimica Acta, vol. 60: 99–101...\..\references\Others\Pashalidis_et_al_1993_Self_Radiolysis_and_Interaction_of_Hypochlorite_with_PuVI.pdf
Pazukhin, E.M., and S.M. Kochergin. 1989. “Stability Constants of Hydrolyzed Forms of Americium(III) and Solubility Product of its Hydroxide.” Soviet Radiochemistry vol. 31: 430–36...\..\references\Others\Pazukhin_Kochergin_1989_Stability_Constants.pdf
Peper, S.M., L.F. Brodnax, S.E. Field, R.A. Zehnder, and S.N. Valdez, and W.H. Runde. 2004. “Kinetic Study of the Oxidative Dissolution of UO2 in Aqueous Carbonate Media.” Industrial Engineering Chemical Research, vol. 43: 8188–93...\..\references\Others\Peper_et_al_2004_Kinetic_Study_of_the_Oxidative_Dissolution.pdf
Popielak, R.S., R.L. Beauheim, S.R. Black, W.E. Coons, C.T. Ellingson, and R.L. Olsen. 1983. Brine Reservoirs in the Castile Formation, Waste Isolation Pilot Plant (WIPP) Project, Southeastern New Mexico. TME-3153. ERMS 242085. Carlsbad, NM: U.S. Department of Energy...\..\references\Others\Popielak_et_al_1983_Brine_Reservoirs_in_the_Castile_Formation_TME3153.pdf
Powers, D.W., S.J. Lambert, S.E. Shaffer, I.R. Hill, and W.D. Weart, eds. 1978. Geological Characterization Report, Waste Isolation Pilot Plant (WIPP) Site, Southeastern New Mexico. SAND78-1596. 2 vols. ERMS 205448. Albuquerque: Sandia National Laboratories...\..\references\Others\Powers_et_al_1978_Geological_Characterization_Report_SAND78_1596.pdf
Pryke, D.C., and J.H. Rees. 1986. “Understanding the Behaviour of the Actinides Under Disposal Conditions: A Comparison between Calculated and Experimental Solubilities.” Radiochimica Acta, vol. 40: 27–32...\..\references\Others\Pryke_Rees_1986_Understanding_Behavior_of_Actinides.pdf
Qui, S. R., C. Amrhein, M.L. Hunt, R. Pfeffer, B. Yakshinskiy, L. Zhang, T.E. Madey, and J.A. Yarmoff. 2001. “Characterization of Uranium Oxide Thin Films Grown from Solution onto Fe Surfaces.” Applied Surface Science, vol. 181: 211–34...\..\references\Others\Qiu_et_al_2001_Characterization_of_U_Oxide_Thin_Films.pdf
Rabindra, N.R., K.M. Vogel, C.E. Good, W.B. Davis, L.N. Roy, D.A. Johnson, A.R. Felmy, and K.S. Pitzer. 1992. “Activity Coefficients in Electrolyte Mixtures: HCl + ThCl4 + H2O for 5–55°C.” Journal of Physical Chemistry, vol. 96: 11,065–072...\..\references\Others\Rabindra_et_al_1992_Activity_Coefficients_in_HCl_ThCl4_H2O_Mixture.pdf
Rai, D., R.G. Strickert, and G.L. McVay. 1982. “Neptunium Concentrations in Solutions Contacting Actinide-Doped Glass.” Nuclear Technology, vol. 58: 69–76...\..\references\Others\Rai_Srickert_McVay_1982_Np_Conctration_in_An_Doped_Glass.pdf
Rai, D., and J.L. Ryan. 1982. “Crystallinity and Solubility of Pu(IV) Oxide and Hydrous Oxide in Aged Aqueous Suspensions.” Radiochimica Acta, vol. 30: 213–16...\..\references\Others\Rai_Ryan_1982_Crystallinity_and_Solubility_of_PuIV_Oxides_in_Aged_Aq_Suspensions.pdf
Rai, D., J.L. Ryan, D.A. Moore, and R.G. Strickert. 1983. “Am(III) Hydrolysis Constants and Solubility of Am(III) Hydroxide.” Radiochimica Acta, vol. 33: 201–06...\..\references\Others\Rai_et_al_1983_AmIII_Hydrolysis_Constants_and_Solubility_of_AmIII_Hydroxide.pdf
Rai, D., and J.L. Ryan. 1985. “Neptunium (IV) Hydrous Oxide Solubility under Reducing and Carbonate Conditions.” Inorganic Chemistry, vol. 24: 247–51...\..\references\Others\Rai_Ryan_1985_NpIV_Hydrous_Oxide_Solubility_under_Reducing_and_Carbonate_Conditions.pdf
Rai, D., A.R. Felmy, and J.L. Ryan. 1990. “Uranium (VI) Hydrolysis Constants and Solubility Products of UO2×xH2O (am).” Inorganic Chemistry, vol. 29: 260–64...\..\references\Others\Rai_Felmy_Ryan_1990_UVI_Hydrolysis_Constants_and_Solubility_Products.pdf
Rai, D., A.R. Felmy, and R.W. Fulton. 1995. “Nd3+ Am3+ Ion Interaction with Sulfate Ion and Their Influence on NdPO4(c) Solubility.” Journal of Solution Chemistry, vol. 24: 879–95...\..\references\Others\Rai_Felmy_Fulton_1995_Nd3_and_Am3_Ion_Interactions_with_Sulfate.pdf
Rai, D., A.R. Felmy, S.M. Sterner, D.A. Moore, M.J. Mason, and C.F. Novak. 1997. “The Solubility of Th(IV) and U(IV) Hydrous Oxides in Concentrated NaCl and MgCl2 Solutions.” Radiochimica Acta, vol. 79: 239–47...\..\references\Others\Rai_et_al_1997_Solubility_of_ThIV_and_UIV.pdf
Rai, D., A.R. Felmy, N.J. Hess, and D.A. Moore. 1998. “A Thermodynamic Model for the Solubility of UO2(am) in the Aqueous K+-Na+-HCO3--CO3 2--OH--H2O System.” Radiochimica Acta, vol. 82: 17–25...\..\references\Others\Rai_Felmy_Hess_Moore_1998_Thermodynamic_Model_for_UO2am_Solubility.pdf
Rai, D., D.A. Moore, C.S. Oakes, and M. Yui. 2000. “Thermodynamic Model for the Solubility of Thorium Dioxide in the Na+-Cl–-OH–-H2O System at 23 °C and 90°C.” Radiochimica Acta, vol. 88: 297–306...\..\references\Others\Rai_et_al_2000_Thermod_Model_for_ThO2_Solub_in_Na_Cl_OH_H2O_System.pdf
Rao, L., D. Rai, A.R. Felmy, R.W. Fulton, and C.F. Novak. 1996. “Solubility of NaNd(CO3)2×6H2O(c) in Concentrated NaCO3 and NaHCO3 Solutions.” Radiochimica Acta, vol. 75: 141–47...\..\references\Others\Rao_et_al_1996_Solubility_of_NaNdCO32_6H2O_in_Concentrated_Carbonate_Solution.pdf
Reed, D.T., S. Okajima, L.H. Brush, and M.A. Molecke. 1993. “Radiolytically-Induced Gas Production in Plutonium-Spiked WIPP Brine.” Materials Research Society Symposium Proceedings (pp. 431-38). Vol. 294. Warrendale, PA: Materials Research Society...\..\references\Others\Reed_et_al_1993_Radiolytically_Induced_Gas_Production.pdf
Reed, D.T., S. Okajima, and M.K. Richmann. 1994. “Stability and Speciation of Plutonium(VI) in WIPP Brine.” Radiochimica Acta, vol. 66/67: 95–101...\..\references\Others\Reed_Okajima_Richmann_1994_Stability_of_PuVI_in_Selected_WIPP_Brines.pdf
Reed, D.T., and D.R. Wygmans. 1997. Actinide Stability/Solubility in Simulated WIPP Brines (March 21). WPO44625. Argonne, IL: Argonne National Laboratory, Actinide Speciation and Chemistry Group, Chemical Technology Group...\..\references\Others\Reed_Wygmans_Richmann_Actinide_Stability_Solubility.pdf
Reed, D.T., S.B. Aase, D. Wygmans, and J. E. Banaszak. 1998. “The Reduction of Np(VI) and Pu(VI) by Organic Chelating Agents.” Radiochimica Acta, vol. 82: 109-14...\..\references\Others\Reed_et_al_1998_Reduction_of_NpVI_and_PuVI_by_Organic_Chelating_Agents.pdf
Reed, D.T., J.F. Lucchini, S.B. Aase, and A.J. Kropf. 2006. “Reduction of Plutonium (VI) in Brine under Subsurface Conditions.” Radiochimica Acta, vol. 94: 591–97...\..\references\Others\Reed_Lucchini_Aase_Kropf_2006_Reduction_of_PuVI_in_Brine.pdf
Reed, D.T., S.E. Pepper, M.K. Richmann, G. Smith, R. Deo, and B.E. Rittmann. 2007. “Subsurface Bio-Mediated Reduction of Higher-Valent Uranium and Plutonium.” Journal of Alloys and Compounds, vol. 444/445: 376–82...\..\references\Others\Reed_Pepper_et_al_2007_Subsurface_Bio_Mediated_Reduction_U_and_Pu.pdf
Reed, D.T., J.F. Lucchini, M. Borkowski, and M.K. Richmann. 2009. Pu(VI) Reduction by Iron under WIPP-Relevant Conditions: Data Summary and Recommendations. LCO-ACP-09, LANL\ACRSP Report. Los Alamos, NM: Los Alamos National Laboratory...\..\references\Others\LCO_ACP_09.pdf
Refait, Ph., and J.-M.R. Génin. 1994. “The Transformation of Chloride-Containing Green Rust One into Sulphated Green Rust Two by Oxidation in Mixed Cl- and SO42- Aqueous Media.” Corrosion Science, vol. 36, no. 1: 55–65...\..\references\Others\Refait_Genin_1994_Transformation_of_Green_Rust_I_Chloride_Into_Green_Rust_II_Sulfate.pdf
Richmann, M.K. 2008. Letter to D. Reed (Subject: Eh/pH Diagrams for Am(III), Th(IV) and Np(V) Based on the FMT Database and Current PA Assumptions). 21 November 2008. U.S. Department of Energy, Los Alamos National Laboratory, Carlsbad Operations, Carlsbad, NM...\..\references\Others\Richmann_to_Reed_2008_Eh_Ph_Diagrams.pdf
Rittmann, B.E., J.E. Banaszak, and D.T. Reed. 2002. “Reduction of Np(V) and Precipitation of Np(IV) by an Anaerobic Microbial Consortium.” Biodegradation, vol. 13: 329–42...\..\references\Others\Rittmann_Banaszak_Reed_2002_Reduction_NpV_and_Precipitation_NpIV.pdf
Rösch, F., T. Reimmann, G.V. Buklanov, M. Milanov, V.A. Khalin, and R. Dryer. 1989. “Electromigration of Carrier-Free Radionuclides. 13. Ion Mobilities and Hydrolysis of 241Am(III) in Aqueous Inert Electrolytes.” Journal of Radioanalytical Nuclear Chemistry, vol. 134: 109–28...\..\references\Others\Rosch_et_al_1989_Electromigration_of_Carrier_Free_Radionuclides.pdf
Runde, W., and J.I. Kim. 1994. Untersuchungen der Übertragbarkeit von Labordaten natürliche Verhältnisse. Chemisches Verhalten von drei- und fünfwertigem Americium in salinen NaCl-Lösungen. Report RCM-01094, Munich: Institut für Radiochemie, Technische Universität München. (Available from National Technical Information Service, 555 Port Royal Road, Springfield, VA, 22161, 703/487-4650 as DE 95752244.)..\..\references\Others\Report_RCM_01094.pdf
Runde, W. 2000: “The Chemical Interactions of Actinides in the Environment.” Los Alamos Science, no. 26: 330–411...\..\references\Others\Runde_2000_Chemical_Interactions_of_Actinides_in_Environment.pdf
Runde, W., S.D. Conradson, D.W. Efurd, N. Lu, D.E. VanPelt, and C.D. Tait. 2002. “Solubility and Sorption of Redox-Sensitive Radionuclides (Np,Pu) in j-13 Water from the Yucca Mountain Site: Comparison Between Experiment and Theory.” Applied Geochemistry, vol. 17: 837–53...\..\references\Others\Runde_et_al_2002_Solubility_and_soprtion_of_Np_and_Pu_in_Yucca_Mountain_Water.pdf
Ryan, J.L, and D. Rai. 1983. “The Solubility of Uranium (IV) Hydrous Oxide in Sodium Hydroxide Solutions under Reducing Conditions.” Polyhedron, vol. 2: 947–52...\..\references\Others\Ryan_Rai_1983_Solubility_of_UIV_Hydrous_Oxide_in_NaOH_under_Reducing_Conditions.pdf
Sanchez, A.L., J.W. Murray, and T.H. Sibley. 1985. “The Adsorption of Plutonium IV and V on Geothite.” Geochimica Cosmochimica Acta, vol. 49: 2297–2307...\..\references\Others\Sanchez_Murray_Sibley_1985_The_Adsorption_of_PuIV_and_PuV_on_Goethite.pdf
Sanchez, L.C., and H.R. Trellue. 1996. Memorandum to T. Hicks (Subject: Estimation of Maximum RH-TRU Thermal Heat Load for WIPP). 17 January 1996. WPO 31165. U.S. Department of Energy, Sandia National Laboratories, Albuquerque, NM...\..\references\Others\Sanchez_Trellue_1996_January_17_Estimation_of_Maximum_RH_TRU_ERMS231165.pdf
Sandino, M.C.A., and B. Grambow. 1994. “Solubility Equilibria in the U(VI)-Ca-K-Cl-H2O System: Transformation of Schoepite into Becquerelite and Compreignacite.” Radiochim. Acta, vol. 66/67: 37-43...\..\references\Others\Sandino_Grambow_1994_Equilibria_UVI_Ca_K_Cl_Transform_Schoepite_To_Becquerelite.pdf
Santschi, P.H., K.A. Roberts, and L. Guo. 2002. “Organic Nature of Colloidal Actinides Transported in Surface Water Environments.” Environmental Science and Technology, vol. 36: 3711–19...\..\references\Others\Santschi_Roberts_Guo_2002_Organic_Nature_of_Colloidal_An_Transported_in_Surface_Water_Environments.pdf
Schuessler, W., B. Kienzler, S. Wilhelm, V. Neck, and J.I. Kim. 2001. Modeling of Near Field Actinide Concentrations in Radioactive Waste Repositories in Salt Formations: Effect of Buffer Materials. Materials Research Society Symposium Proceedings, vol. 663: 791-98...\..\references\Others\Schuessler_et_al_2001_Modeling_An_Concentrations_in_Salt_Bed_Waste_Repository.pdf
Shannon, R.D. 1976. “Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides.” Acta Cryst, vol. A32: 751–67...\..\references\Others\Shannon_1976_Revised_Effective_Ionic_Radii_and_Systematic_Studies_of_Interatomic_Distances.pdf
Siekierski, S. 1988. “Comparison of Yttrium, Lanthanides and Actinides in Respect to Unit Cell Volumes of Isostructural Compounds and Thermodynamic Functions of Complex Formation.” Journal of Radioanalytical Nuclear Chemistry, vol. 122: 279–84...\..\references\Others\Siekierski_1988_Comparison_of_Yttrium_Lanthanides_and_Actinides_in_Respect_to_Unit_Cell_Volumes.pdf
Silva, R.J., G. Bidoglio, M.H. Rand, P.B. Robouch, H. Wanner, and I. Puigdomenech. 1995. Chemical Thermodynamics of Americium. Chemical Thermodynamics Series 2. New York Elsevier...\..\references\Others\Silva_et_al_1995_Chemical_Thermodynamics_of_Americium.pdf
Skokan, C.K., M.C. Pfeifer, G.V. Keller, and H.T. Andersen. 1987. Studies of Electrical and Electromagnetic Methods for Characterizing Salt Properties at the WIPP Site, New Mexico. SAND87-7174. Carlsbad NM: Sandia National Laboratories...\..\references\Others\Skokan_Pfeifer_Keller_Andersen_1987_Studies_of_Electrical_and_Electromagnetic_Methods_SAND87_7174.pdf
Snider, A.C. 2003a. Verification of the Definition of Generic Weep Brine and the Development of a Recipe for This Brine. ERMS 527505. Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Snider_2003_Verification_of_the_Definition_of_GWB_and_Recipe_Development_ERMS527505.pdf
Snider, A.C. 2003b. “Hydration of Magnesium Oxide in the Waste Isolation Pilot Plant.” Materials Research Society Symposium Proceedings (pp. 665–70). Vol. 757. Warrendale, PA: Materials Research Society...\..\references\Others\Snider_2003_Hydration_of_Magnesium_Oxide_in_the_WIPP.pdf
Spinks, J.W.T., and R.J. Woods. 1990. “Radiation Sources: The Interaction of Radiation with Matter.” Introduction to Radiation Chemistry (pp. 243–313). New York: Wiley 1990...\..\references\Others\Spinks_Woods_1990_Radiation_Sources_Interaction_of_Radiation_with_Matter.pdf
Stadler, S., and J.I. Kim. 1988. “Hydrolysis reactions of Am(III) and Am(V).” Radiochim. Acta, 44/45: 39–44...\..\references\Others\Stadler_Kim_1988_Hydrolysis_of_AmIII_and_AmV.pdf
Stein, J.S. 2005. Memorandum to L.H. Brush (Subject: Estimate of Volume of Brine in Repository That Leads to a Brine Release). 13 April 2005. ERMS 539372. U.S. Department of Energy, Sandia National Laboratories, Albuquerque, NM...\..\references\Others\Stein_to_Brush_2005_April_13_Estimate_of_Volume_of_Brine_ERMS539372.pdf
Stoelzel, D.M., and D.G. O’Brien. 1996. Analysis Package for the BRAGFLO Direct Release Calculations (Task 4) of the Performance Assessment Calculations Supporting the Compliance Certification Application (CCA), AP-029, Brine Release Calculations. ERMS 240520. Albuquerque: Sandia National Laboratories...\..\references\Others\Stoelzel_Brien_Analysis_Package_for_the_BRAGFLO_Direct_Release_Calculations_Task_4_ERMS417870.pdf
Telander, M.R., and R.E. Westerman. 1993. Hydrogen Generation by Metal Corrosion in Simulated Waste Isolation Pilot Plant Environments: Progress Report for the Period November 1989 through December 1992. SAND92-7347. ERMS 223456. Albuquerque: Sandia National Laboratories...\..\references\Others\Telander_Westerman_1993_Hydrogen_Generation_by_Metal_Corrosion_SAND92_7347.pdf
Telander, M.R., and R.E. Westerman. 1997. Hydrogen Generation by Metal Corrosion in Simulated Waste Isolation Pilot Plant Environments. SAND96-2538. Albuquerque: Sandia National Laboratories...\..\references\Others\Telander_Westerman_1997_Hydrogen_Generation_by_Metal_Corrosion_SAND96_2538.pdf
Torrero, M.E., I. Casas, J. de Pablo, M.C.A. Sandino, and B.A. Grambow. 1994. “Comparison Between Unirradiated UO2(s) and Schoepite Solubilities in 1 M NaCl Medium.” Radiochimica Acta, vol. 66/67: 29–35...\..\references\Others\Torrero_et_al_1994_Comparison_of_Unirradiated_UO2_and_Schoepite_solubilities_in_1M_NaCl.pdf
Torretto, P., K. Becraft, T. Prussin, K. Roberts, S. Carpenter, D. Hobart, and H. Nitsche. 1995. Solubility and Speciation Results from Oversaturation Experiments on Neptunium, Plutonium and Americium in a Neutral Electrolyte with a Total Carbonate Similar to Water from Yucca Mountain Region Well UE-25p#1. Report LA-13018-MS. Los Alamos: Los Alamos National Laboratory...\..\references\Others\Torretto_et_al_1995_Solubility_and_Speciation_of_Np_Pu_and_Am_LA_13018_MS.pdf
Tremaine, P.R., J.D. Chen, G.J. Wallace, and W.A. Boivin. 1981. “Solubility of Uranium (IV) Oxide in Alkaline Aqueous Solutions to 300°C.” Journal of Solution. Chemistry, vol. 10: 221–30...\..\references\Others\Tremaine_et_al_1981_Solubility_of_UIV_Oxide_in_Alkaline_Solutions.pdf
Trolard, F., J.M.R. Genin, M. Abdelmoula, G. Bourrie, B. Humbert, and A. Herbillon. 1997. “Identification of a Green Rust Mineral in a Reductomorphic Soil by Mossbauer and Raman Spectroscopies.” Geochimica Cosmochimica Acta, vol. 63: 1107–11...\..\references\Others\Trolard_et_al_1997_Identification_of_Green_Rust_in_Soil_by_Mossbauer_and_Raman_Spectroscopies.pdf
U.S. Department of Energy (DOE). 1996. Title 40 CFR Part 191 Compliance Certification Application for the Waste Isolation Pilot Plant (October). 21 vols. DOE/CAO 1996-2184. Carlsbad, NM: Carlsbad Area Office...\..\references\CCA\CCA.htm
U.S. Department of Energy (DOE). 2004. Title 40 CFR Part 191 Compliance Recertification Application for the Waste Isolation Pilot Plant (March). 10 vols. DOE/WIPP 2004-3231. Carlsbad, NM: Carlsbad Field Office...\..\references\CRA-2004\CRA-2004.htm
U.S. Department of Energy (DOE). 2006. Transuranic Waste Baseline Inventory Report—2004(Revision 0). DOE/TRU 2006-3344. Carlsbad, NM: Carlsbad Field Office...\..\references\Others\DOE_2006_TRU_Waste_Baseline_Inventory_Report_2006_3344_full.pdf
U.S. Environmental Protection Agency (EPA). 1993. “40 CFR Part 191: Environmental Radiation Protection Standards for the Management and Disposal of Spent Nuclear Fuel, High-Level and Transuranic Radioactive Wastes; Final Rule.” Federal Register, vol. 58 (December 20, 1993): 66398–416...\..\references\Others\EPA_58_FR_66398_416_December_20_1993.pdf
U.S. Environmental Protection Agency (EPA). 2005. Teleconference with U.S. Department of Energy (DOE), Sandia National Laboratories (SNL), and Los Alamos National Laboratory (LANL) re: Change in U(VI) Solubility Assumption to a Concentration to 1 mM. 2 March 2005...\..\references\Others\EPA_2005_March_2_Teleconference.pdf
U.S. Environmental Protection Agency (EPA). 2006. Technical Support Document for Section 194.24: Evaluation of the Compliance Recertification Actinide Source Term and Culebra Dolomite Distribution Coefficient Values (March). Washington, DC: Office of Radiation and Indoor Air...\..\references\Others\EPA_2006_TSD_194_24_Evaluation_of_the_Compliance_Recertification_Actinide.pdf
Van Luik, A.E., M.J. Apted, W.J. Bailey, J.H. Haberman, J.S. Shade, R.E. Guenther, R.J. Serne, E.R. Gilbert, R. Peters, and R.E. Williford. 1987. Spent Nuclear Fuel as a Waste Form for Geologic Disposal: Assessment and Recommendations on Data and Modeling Needs. PNL-6329. Richland, WA: Pacific Northwest Laboratories...\..\references\Others\Van_Luik_et_al_1987_Assessment_and_Recommendations_on_Data_PNL6329.pdf
Villarreal, R., J.M. Bergquist, and S.L. Leonard. 2001. The Actinide Source-Term Waste Test Program (STTP): Final Report. 3 vols. LA-UR-01-6822, LA-UR-01-6912 and LA-UR-01-6913. Los Alamos: Los Alamos National Laboratory...\..\references\Others\Villarreal_et_al_2001_Actinide_Source_Term_Waste_Test_Vol_I_III.pdf
Wall, N.A., and S.A. Matthews. 2005. “Sustainability of Humic Acids in the Presence of Magnesium Oxide.” Applied Geochemistry, vol. 20: 1704–13...\..\references\Others\Wall_Mathews_2005_Sustainability_of_Humic_Acids_in_the_Presence_of_MgO.pdf
Walther, C. 2003. “Comparison of Colloid Investigations by Single Particle Analytical Techniques: A Case Study on Thorium-Oxyhydroxides.” Colloids and Surfaces A: Physicochemical Engineering Aspects, vol. 217: 81–92...\..\references\Others\Walther_2003_Comparison_of_colloid_investigations.pdf
Wang, Y., and L. Brush. 1996a. Memorandum to M.S. Tierney (Subject: Estimates of Gas-Generation Parameters for the Long-Term WIPP Performance Assessment). 26 January 1996. ERMS 231943. U.S. Department of Energy, Sandia National Laboratories, Albuquerque, NM...\..\references\Others\Wang_and_Brush_to_Tierney_1996_January_26_Estimates_of_Gas_Generation_Parameters_ERMS231943.pdf
Wang, Y., and L. Brush. 1996b. Memorandum to M.S. Tierney (Subject: Modify the Stoichiometric Factory in the BRAGFLO to Include the Effect of MgO Added to WIPP Repository as a Backfill). 23 February 1996. ERMS 232286. U.S. Department of Energy, Sandia National Laboratories, Albuquerque, NM...\..\references\Others\Wang_Brush_to_Tierney_1996_February_23_Modify_the_Stoichiometric_Factory_in_BRAGFLO.pdf
Weiner, R. 1996. Technical memorandum to SWCF-A: Records Center (Subject: Documentation Package For: Oxidation State Distribution of Actinides in the Repository). 27 March 1996. ERMS 235194. U.S. Department of Energy, Sandia National Laboratories, Albuquerque, NM...\..\references\Others\Weiner_to_Records_Center_1996_March_27_Documentation_Package_For_Oxidation_ERMS235194.pdf
White, A.F., A. Yee, and S. Flexser. 1985. “Surface Oxidation-Reduction Kinetics Associated with Experimental Basalt-Water Reactions at 25 ºC.” Chemical Geology, vol. 49: 73...\..\references\Others\White_Yee_Flexser_1985_Surface_Oxidation_Reduction_Kinetics.pdf
Wolery, T.J. 1992. EQ3/6, A Software Package for Geochemical Modeling of Aqueous Systems: Package Overview and Installation Guide(Version 7.0). UCRL-MA-110662 PT 1. Livermore, CA: Lawrence Livermore National Laboratory...\..\references\Others\Wolery_1992_EQ36_A_Software_Package_for_Geochemical_Modeling_of_Aqueous_Systems_ERMS241375.pdf
Wolery, T.J., and S.A. Daveler. 1992. EQ6, A Computer Program for Reaction-Path Modeling of Aqueous Geochemical Systems: Theoretical Manual, User’s Guide, and Related Documentation(Version 7.0). UCRL-MA-110662 PT IV. Livermore, CA: Lawrence Livermore National Laboratory...\..\references\Others\Wolery_Daveler_1992_EQ36_A_Computer_Program_for_Reaction_Path_Modeling_of_Aqueous_Geochemical_Systems_ERMS241379.pdf
Xiong, Y. 2005. E-mail to J.F. Kanney and J.J. Long (Subject: Release of FMT_050405.CHEMDAT). 5 April 2005. ERMS 539304. U.S. Department of Energy, Sandia National Laboratories, Carlsbad, NM...\..\references\Others\Xiong_to_Kanney_Long_2005_Release_of_FMT_ERMS539304.pdf
Xiong, Y., E.J. Nowak, and L.H. Brush. 2005. Updated Uncertainty Analysis of Actinide Solubilities For the Response to EPA Comment C-23-16, Rev. 1 (April 28). ERMS 539595. (supersedes ERMS 538219). Carlsbad, NM: Sandia National Laboratories...\..\references\Others\Xiong_Nowak_Brush_2005_Updated_Uncertainty_Analysis_of_Actinide_Solubilities_ERMS539595.pdf
Xiong, Y., and A.S. Lord. 2008. “Experimental Investigations of the Reaction Path in the MgO-CO2-H2O System in Solution with Various Ionic Strengths, and Their Applications to Nuclear Waste Isolation.” Applied Geochemistry, vol. 23: 1634–59...\..\references\Others\Xiong_Lord_2008_Experimental_Investigations_of_the_Reaction_Path_SAND2006_7185J.pdf
Yajima, T., Y. Kawanura, and S. Ueta. 1995. “Uranium(IV) Solubility and Hydrolysis Constants Under Reduced Conditions.” Materials Research Society Symposium Proceedings, (pp. 1137–42). Vol. 353. Warrendale, PA: Materials Research Society...\..\references\Others\Yajima_Kawamura_Ueta_1995_UIV_Solubility_and_Hydrolysis_Constants.pdf
Yamamura, T., A. Kitamura, A. Fukui, S. Nishikawa, T. Yamamoto, and H. Moriyama. 1998. “Solubility of U(VI) in Highly Basic Solutions.” Radiochimica Acta, vol. 83: 139–146...\..\references\Others\Yamamura_et_al_1998_Solubility_UVI.pdf
Yamazaki, H., B. Lagerman, V. Symeopoulos, and G.R. Choppin. 1992. “Solubility of Uranyl in Brine.” Radioactive Waste Management, vol. 1992: 1607–11...\..\references\Others\Yamazaki_Lagerman_Symeopoulos_Choppin_1992_Solubility_of_Uranyl_in_Brine.pdf
Zavarin, M., A.B. Kersting, P. Zhao, E.R. Sylwester, P.G. Allen, and R.W. Williams. 2003. “Plutonium Colloid-Facilitated Transport in the Environment-Experimental and Transport Modeling: Evidence for Plutonium Migration Mechanisms.” Plutonium Futures—The Science Conference Proceedings (pp. 102–04.), G.D. Jarvinen, ed...\..\references\Others\Zavarin_et_al_2003_Pu_Colloid_Transport.pdf
Zitomer, D.H., and R.E. Speece. 1993. “Sequential Environments for Enhanced Biotransformation of Aqueous Contaminants.” Environmental Science and Technology, vol. 27: 227...\..\references\Others\Zitomer_Speece_1993_Enhanced_Biotransformation_of_Aqueous_Contaminants.pdf
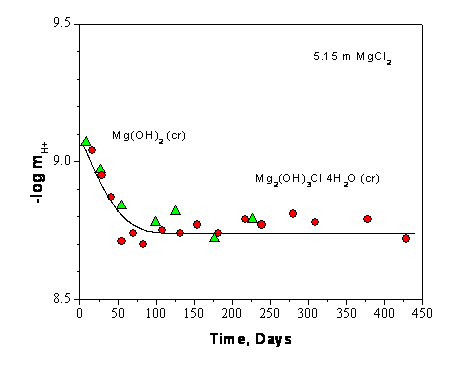

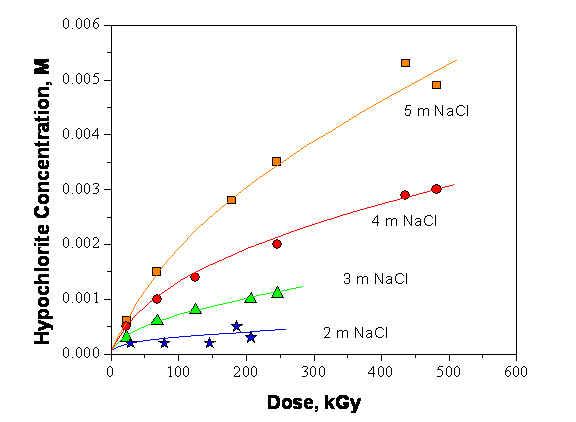
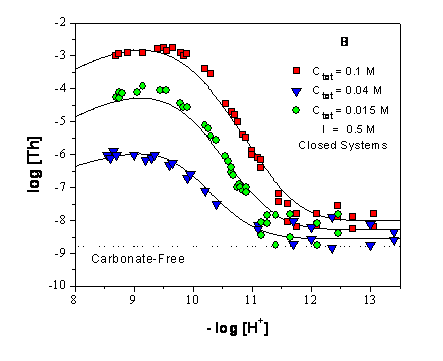
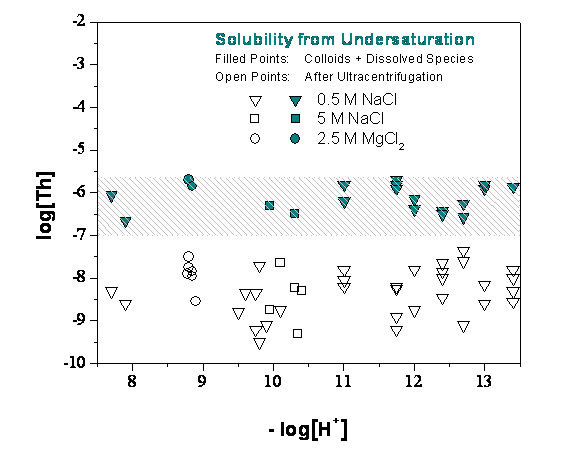

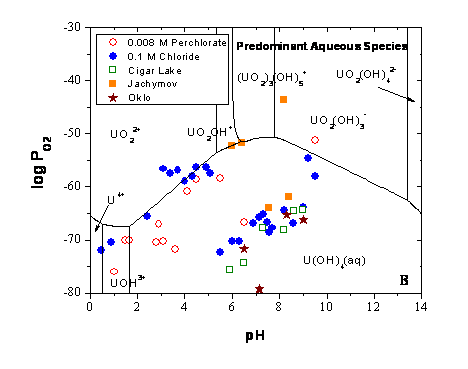
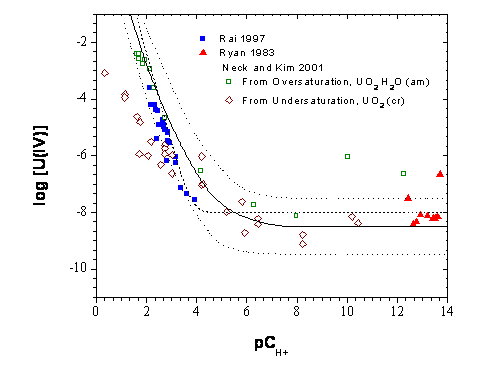
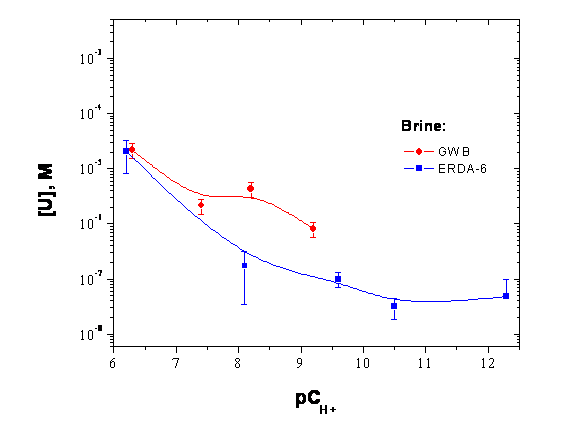
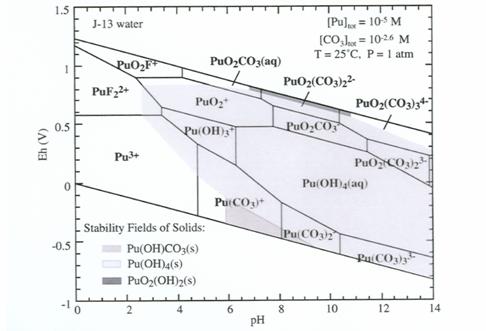
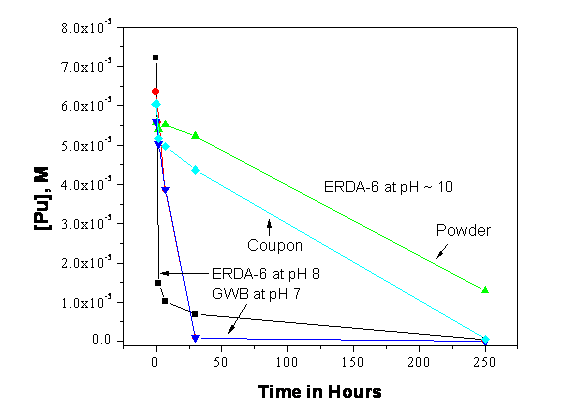
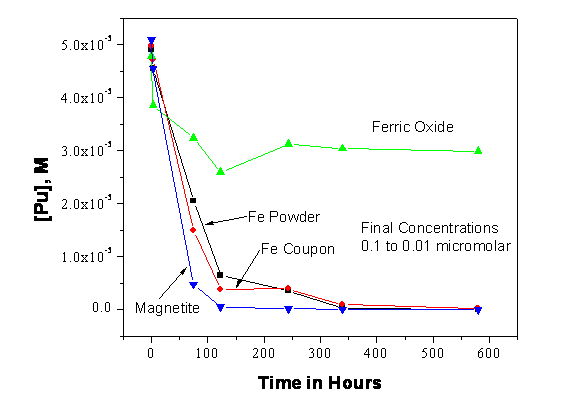
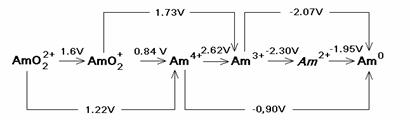
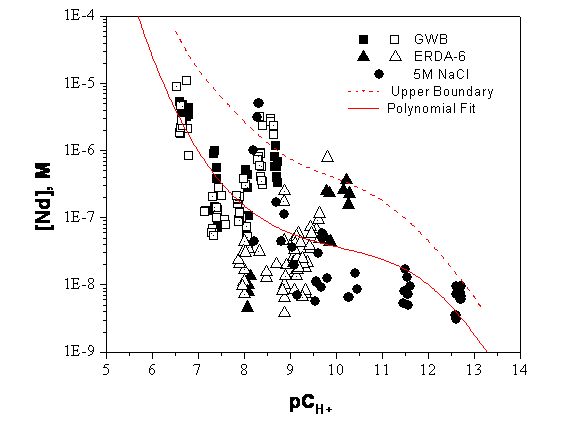
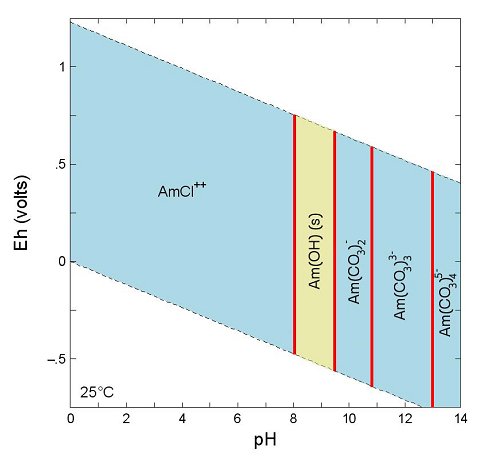
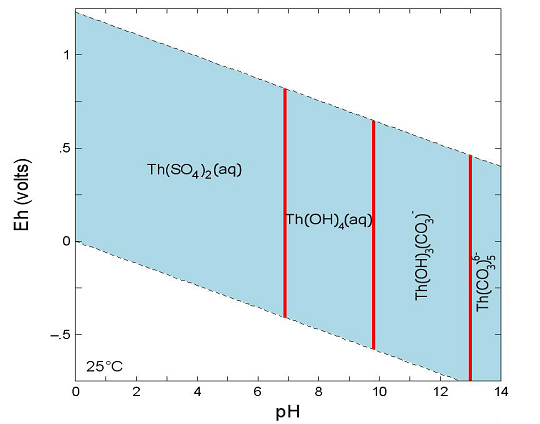
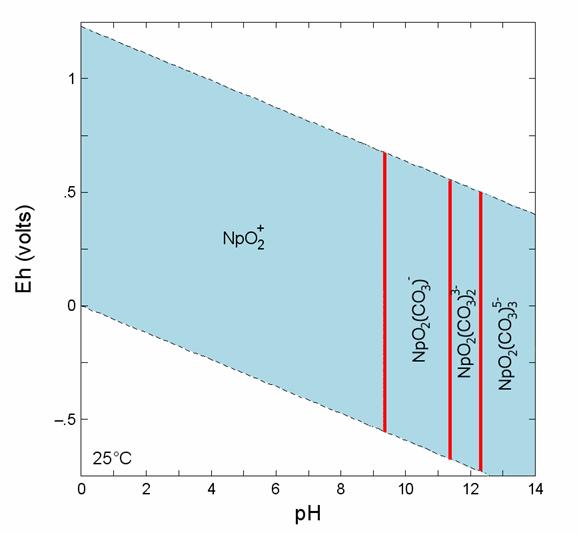
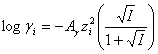 (
(